

I punti caldi della mutazione SARS-CoV-2 emergenti includono una nuova variante della RNA polimerasi RNA-dipendente
Articolo Originale: BMC Journal of Translational Medicine
Traduzione: The Solver Team
Astratto
sfondo
SARS-CoV-2 è un coronavirus a RNA responsabile della pandemia della sindrome respiratoria acuta grave (COVID-19). I virus a RNA sono caratterizzati da un alto tasso di mutazione, fino a un milione di volte superiore a quello dei loro ospiti. La capacità mutagena del virus dipende da diversi fattori, inclusa la fedeltà degli enzimi virali che replicano gli acidi nucleici, come la RNA polimerasi RNA dipendente da SARS-CoV-2 (RdRp). La velocità di mutazione guida l’evoluzione virale e la variabilità del genoma, consentendo così ai virus di sfuggire all’immunità dell’ospite e di sviluppare resistenza ai farmaci.
Metodi
Abbiamo analizzato 220 sequenze genomiche dal database GISAID derivate da pazienti infetti da SARS-CoV-2 in tutto il mondo da dicembre 2019 a metà marzo 2020. Il genoma di riferimento di SARS-CoV-2 è stato ottenuto dal database GenBank. L’allineamento dei genomi è stato eseguito utilizzando Clustal Omega. I test di Mann-Whitney e Fisher-Exact sono stati utilizzati per valutare la significatività statistica.
Risultati
Abbiamo caratterizzato 8 nuove mutazioni ricorrenti di SARS-CoV-2, situate nelle posizioni 1397, 2891, 14408, 17746, 17857, 18060, 23403 e 28881. Le mutazioni nelle posizioni 2891, 3036, 14408, 23403 e 28881 sono prevalentemente osservate in Europa, mentre quelli situati nelle posizioni 17746, 17857 e 18060 sono presenti esclusivamente in Nord America. Abbiamo notato per la prima volta una mutazione silente nel gene RdRp in Inghilterra (Regno Unito) il 9 febbraio 2020 mentre una diversa mutazione in RdRp che modifica la sua composizione amminoacidica è emersa il 20 febbraio 2020 in Italia (Lombardia). I virus con mutazione RdRp hanno una mediana di 3 mutazioni puntiformi [range: 2–5], altrimenti hanno una mediana di 1 mutazione [range: 0–3] (valore p <0,001).
Conclusioni
Questi risultati suggeriscono che il virus si sta evolvendo e che ceppi europei, nordamericani e asiatici potrebbero coesistere, ciascuno caratterizzato da un diverso modello di mutazione. Il contributo del mutato RdRp a questo fenomeno deve essere studiato. Ad oggi, diversi farmaci mirati agli enzimi RdRp vengono impiegati per il trattamento dell’infezione da SARS-CoV-2. Alcuni di loro hanno una porzione di legame prevista in una fessura idrofobica SARS-CoV-2 RdRp, che è adiacente alla mutazione 14408 che abbiamo identificato. Di conseguenza, è importante studiare e caratterizzare la mutazione RdRp di SARS-CoV-2 al fine di valutare possibili fenotipi virali di resistenza ai farmaci. È anche importante riconoscere se la presenza di alcune mutazioni potrebbe essere correlata a diversi tassi di mortalità per SARS-CoV-2.
sfondo
La recente comparsa in Cina del nuovo patogeno umano, la sindrome respiratoria acuta grave, Coronavirus 2 (SARS-CoV-2), e la sua rapida diffusione a livello nazionale e internazionale, rappresentano un’emergenza sanitaria globale. L’11 marzo 2020, l’OMS ha dichiarato pubblicamente l’epidemia di SARS-CoV-2 come una pandemia. In poche settimane, il virus ha causato migliaia di morti in tutto il mondo, con un forte impatto sull’economia globale e sulle abitudini umane. SARS-CoV-2 è un virus con involucro + ssRNA, appartenente al genere Betacoronavirus che include altri due virus a RNA che hanno causato recenti importanti epidemie: la sindrome respiratoria acuta grave (SARS) causata da SARS-CoV e la sindrome respiratoria del Medio Oriente (MERS) di MERS-CoV.
Degno di nota, alcune prove sono state fornite di recente, a sostegno del fatto che la mortalità per SARS-CoV-2 può differire significativamente a seconda dell’area geografica. Ad esempio, Baud e colleghi hanno riferito che il tasso di mortalità è tre volte superiore in Cina (15,2% [IC 95% 12,5-17,9] in Cina, rispetto al 5,6% [IC 95% 5,4-5,8] in Cina) [ 1 ] . Questo tasso è stato rivalutato dividendo il numero di decessi in un dato giorno per il numero di pazienti con infezione confermata da SARS-CoV-2 14 giorni prima, considerando i dati dell’OMS relativi al numero cumulativo di decessi al 1 ° marzo 2020 [ 1]. Le differenze nei tassi di infezione virale possono essere dovute a una combinazione di fattori, comprese le diverse strategie nazionali adottate per le limitazioni di movimento delle persone, l’isolamento e la quarantena, la diversa immunità genetica della popolazione di gregge. Le differenze di mortalità devono essere comprese, ma le mutazioni virali e la capacità di evoluzione nel tempo possono essere importanti.
Il tasso di mutazione dei virus a RNA è drammaticamente alto, fino a un milione di volte superiore a quello dei loro ospiti e questo alto tasso è correlato alla modulazione della virulenza e all’evolvibilità, tratti considerati benefici per l’adattamento virale [ 2 ]. Wang e colleghi hanno recentemente caratterizzato 13 siti di variazione nelle regioni SARS-CoV-2 ORF1ab, S, ORF3a, ORF8 e N, tra cui le posizioni 28144 in ORF8 e 8782 in ORF1a hanno mostrato un tasso di mutazione del 30,53% e 29,47%, rispettivamente [ 3 ]. I risultati riportati in precedenza mostrano che SARS-CoV-2 si sta rapidamente spostando attraverso i paesi e stanno emergendo genomi con nuovi hotspot di mutazione.
Il tasso di mutazione del virus RNA contribuisce all’adattamento virale creando un equilibrio tra l’integrità delle informazioni genetiche e la variabilità del genoma [ 4 , 5 , 6]. La caratterizzazione biologica delle mutazioni virali può fornire preziose informazioni per valutare la resistenza ai farmaci virali, la fuga immunitaria e i meccanismi correlati alla patogenesi. Inoltre, gli studi sulle mutazioni virali possono essere cruciali per la progettazione di nuovi vaccini, farmaci antivirali e test diagnostici. Il processo mutageno del genoma virale dipende dagli enzimi virali che replicano gli acidi nucleici, influenzati da poche o nessuna capacità di correzione di bozze e / o riparazione degli acidi nucleici post-replicativa. Altri processi generatori di mutazioni includono: enzimi ospiti, danni spontanei agli acidi nucleici dovuti a mutageni fisici e chimici, eventi di ricombinazione e anche particolari elementi genetici responsabili della produzione di nuove varianti. I tassi di mutazione sono modulati da altri fattori come i determinanti della sequenza e della struttura del modello coinvolte nella replicazione virale.
Le RNA polimerasi RNA-dipendenti (RdRps) sono proteine multi-dominio in grado di catalizzare la formazione dipendente dal modello RNA di legami fosfodiestere tra ribonucleotidi in presenza di ioni metallici bivalenti [ 7 , 8 , 9 ]. Nella maggior parte dei virus, la RNA polimerasi manca di capacità di correzione di bozze, con alcune eccezioni come l’ ordine Nidovirales (a cui appartiene il genere Coronavirus ), che si distingue per avere i genomi di RNA più grandi. I nidovirus sono caratterizzati da un complesso macchinario dedicato alla sintesi dell’RNA, cioè azionato da proteine non strutturali (nsps), prodotte come prodotti di scissione delle poliproteine virali ORF1a e ORF1b [ 10] per facilitare la replicazione e la trascrizione del virus.
Il SARS-CoV-2 RdRp (denominato anche nsp12) è un componente chiave del meccanismo di replicazione / trascrizione. SARS-CoV-2 condivide un’alta omologia per nsp12 rispetto a SARS-CoV, suggerendo che la sua funzione e il suo meccanismo d’azione potrebbero essere ben conservati [ 11 ]. Ciò è stato confermato da un recente studio strutturale cryo-EM ottenuto per SARS-CoV-2 nsp12 [ 12 ]. In SARS-CoV, è stata segnalata un’attività esonucleasica con funzione di correzione di bozze per nsp14 (ExoN), e anche una proteina nsp14 omologa si trova nel SARS-CoV-2 [ 11 , 13 ]. ExoN aumenta la fedeltà della sintesi dell’RNA correggendo gli errori di incorporazione dei nucleotidi effettuati da RdRp [ 14]. L’inattivazione genetica del coronavirus ExoN si traduce in una diminuzione di 21 volte nella fedeltà di replicazione rispetto a SARS-CoV wild-type [ 15 ]. Inoltre, Kirchdoerfer e colleghi hanno mostrato il coinvolgimento di nsp7 e nsp8 nella formazione di un supercomplesso con RdRp in SARS-CoV [ 16 ], e questo è stato confermato anche per SARS-CoV-2 in un recente studio che svela la struttura di SARS- Complesso CoV-2 RdRp / nsp7 / nsp8 [ 12 ]. Questo complesso garantisce la processività RdRp, diventando fondamentale nella fedeltà della trascrizione. Tuttavia, i residui critici SARS-CoV RdRp coinvolti nell’interazione ExoN, nsp7 e nsp8 devono ancora essere identificati.
Gli RdRps sono considerati tra gli obiettivi primari per lo sviluppo di farmaci antivirali, contro un’ampia varietà di virus. Alcuni inibitori RdRp sono stati considerati come bersaglio di SARS-CoV-2: Favipiravir [ 17 ], Galidesivir [ 18 ], Remdesivir [ 19 ] e Ribavirina [ 20 ]. È interessante notare che il sito di attracco non si trova in prossimità del dominio catalitico del RdRp [ 21 ]. Inoltre, si prevede che altri possibili farmaci come Filibuvir, Cepharanthine, Simeprevir e Tegobuvir siano potenziali inibitori di RdRp [ 22 ]. Mutazioni che si verificano naturalmente in residui critici per l’efficacia del farmaco possono portare a fenomeni di farmacoresistenza, con una significativa perdita dell’affinità di legame di queste molecole con il RdRp.
Abbiamo concentrato il nostro studio sulle mutazioni della SARS-CoV-2 al fine di valutare se nuove varianti virali si stessero diffondendo nei Paesi. Questa caratterizzazione delle varianti SARS-CoV-2 potrebbe portare a migliori trattamenti terapeutici, progettazione di vaccini e approcci diagnostici.
Metodi
La sequenza di riferimento del virus SARS-CoV-2 utilizzata per l’analisi è stata depositata nel gennaio 2020 da Wu e colleghi [ 11 ] precedentemente chiamata “Wuhan Seafood market pneumonia virus” (WSM, NC_045512) ( https: //www.ncbi.nlm.nih .gov / nuccore / NC_045512 ) . Database GISAID ( https://www.gisaid.org/) filtrato da dicembre 2019 fino al 13 marzo 2020 è stato utilizzato per raccogliere 220 genomi completi SARS-CoV-2 di diversi pazienti in tutto il mondo (ad esempio Cina, Stati Uniti, Canada, Australia, Regno Unito, Germania, Francia, Giappone, Italia , Svizzera, Singapore, Lussemburgo, Paesi Bassi, Spagna, Portogallo, Svezia, Repubblica Ceca, Thailandia, India, Cambogia, Hong Kong, Finlandia, Singapore e Irlanda) prendendo in particolare considerazione quelli depositati durante lo sviluppo dei focolai europei. Sono stati analizzati solo genomi completi (28.000-30.000 bps).
Gli strumenti Clustal Omega, Serial Cloner e Blast sono stati utilizzati per eseguire l’allineamento di sequenze multiple, confrontando la sequenza WSM con le sequenze isolate dai pazienti, mentre Swiss Model ed Ez-mol sono stati utilizzati per la modellazione delle proteine.
L’analisi statistica è stata eseguita dal software R. Abbiamo prima verificato la normalità della distribuzione dei dati con il test di Shapiro – Wilk, esprimendo le variabili continue come mediana e range (min – max). Le variabili categoriali sono state espresse come frequenza e percentuali assolute. I test non parametrici di Mann-Whitney e Fisher-Exact sono stati utilizzati per confrontare il numero di mutazioni per genoma con almeno una delle mutazioni selezionate rispetto al gruppo di genomi che non presentano la specifica mutazione analizzata. Tutti i valori di p sono stati calcolati da test a due code utilizzando 0,05 come livello di significatività.
Risultati
Identificazione di punti critici di mutazione in diverse aree geografiche
Un database di 220 sequenze genomiche complete SARS-CoV-2 isolate dal paziente raccolte in modo casuale dal database GISAID è stato allineato e confrontato con il genoma di riferimento WSM SARS-CoV-2. In particolare, 5 genomi isolati da pazienti sono stati inviati al database GISAID a dicembre 2019 (2,3%), 67 a gennaio 2020 (30,45%), 67 a febbraio 2020 (30,45%) e 81 (36,8%) fino al 13 Marzo 2020. Circa il 33,6% dei genomi completi appartiene a pazienti di età inferiore a 44 anni, che è l’età media dei pazienti inclusi nel database. La maggior parte dei pazienti sono uomini (55,5%).
Abbiamo diviso il nostro set di dati in 4 aree geografiche: Asia, Oceania, Europa, Nord America (Fig. 1 ). All’interno di ciascuna area abbiamo eseguito analisi di allineamento confrontando i genomi dei pazienti con la sequenza di riferimento. Il gruppo asiatico comprende genomi ottenuti da pazienti situati in Cina, Giappone, Sud-est asiatico e India. Il gruppo oceaniano comprende genomi di pazienti australiani, mentre quello europeo comprende ogni genoma ottenuto da pazienti situati in ciascuno degli stati europei (Spagna, Portogallo, Regno Unito, Paesi Bassi, Italia, Germania, Svizzera, Francia, Lussemburgo, Svezia, Finlandia , Danimarca e Belgio). Infine, il gruppo del Nord America contiene genomi di pazienti statunitensi e canadesi.
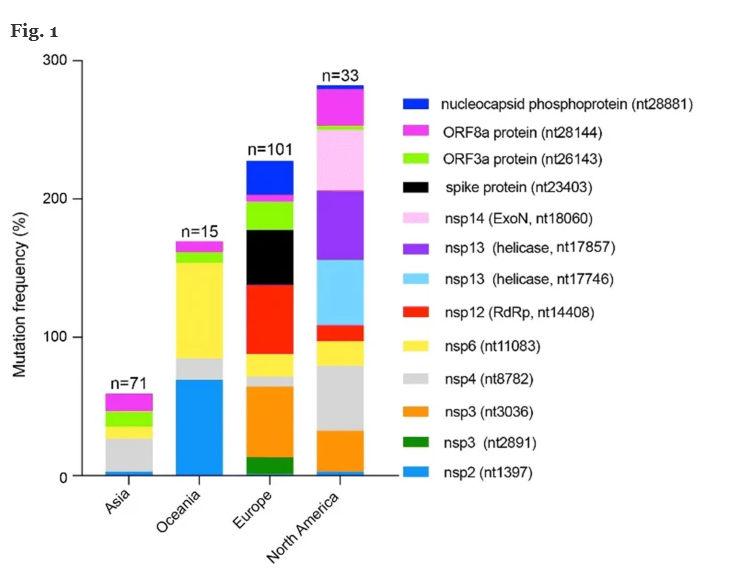
Abbiamo valutato la distribuzione di SARS-CoV-2 mutazioni attraverso diverse aree geografiche (vedi Fig. 1 ), il calcolo della frequenza di mutazione in queste 4 aree geografiche, normalizzando il numero di genomi trasportano una determinata mutazione per area geografica.
Abbiamo confermato il verificarsi di mutazioni situate nelle posizioni 3036, 8782, 11083, 28144 e 26143 [ 23 , 24 , 25 , 33 ]. Inoltre, abbiamo evidenziato la presenza di ulteriori “mutazioni conservate” in tutte le aree geografiche, tenendo conto solo di quelle occorse più di 10 volte nel nostro database. Quelli con un’incidenza inferiore non sono stati segnalati. Queste mutazioni sono state trovate in posizione 1397, 2891, 14408, 17746, 17857, 18060, 23403, 28881, appartenenti a ORF1ab (1397 nsp2, 2891 nsp3, 14408 RdRp, 17746 e 17857 nsp143, 18060 nsp14), S (23403, spike protein ) e ORF9a (28881, fosfoproteina nucleocapside), rispettivamente.
Abbiamo scoperto che 3 delle 12 mutazioni più frequenti (posizioni 3036, 8782 e 18060) erano silenti, mentre una mutazione (posizione 11083) era al di fuori della sequenza ORF. D’altra parte, le mutazioni 1397, 2891, 14408, 17746, 17857, 23403, 26143, 28144 e 28881 hanno determinato le seguenti modifiche degli amminoacidi: 1397 (da V a I), 14408 (da P a L), 17746 (da P a L) ), 17857 (da C a Y), 23403 (da D a G), 26143 (da G a V), 28144 (da L a S). La mutazione situata nella posizione 28881 è correlata a una mutazione del doppio codone, che induce la sostituzione di due amminoacidi, vale a dire 28881 (da R a K) e (G a R). Il nuovo amminoacido presente nel 1397 (da V a I), 14408 (da P a L), 17746 (da P a L), 17857 (da C a Y), 26143 (da G a V) e 28144 (da L a S) aveva un punto isoelettrico rispetto all’amminoacido originale presente nelle sequenze proteiche di riferimento,ad eccezione delle mutazioni alle posizioni 23403 (da D a G), 28881 (da R a K) e 28881 (da G a R), dove l’amminoacido mutato ha un punto isoelettrico significativamente diverso. Sono necessari ulteriori studi per determinare se queste mutazioni hanno un impatto sulla funzione e sulla struttura delle proteine. Abbiamo notato che il numero e il verificarsi di ciascuna mutazione aumenta nei genomi rilevati in Asia, raggiungendo un massimo nei genomi trovati in Europa e Nord America. Abbiamo anche notato che i ceppi virali trovati in Europa e Nord America derivano dal “ceppo” L- originario dell’Asia [Abbiamo notato che il numero e il verificarsi di ciascuna mutazione aumenta nei genomi rilevati in Asia, raggiungendo un massimo nei genomi trovati in Europa e Nord America. Abbiamo anche notato che i ceppi virali trovati in Europa e Nord America derivano dal “ceppo” L- originario dell’Asia [Abbiamo notato che il numero e il verificarsi di ciascuna mutazione aumenta nei genomi rilevati in Asia, raggiungendo un massimo nei genomi trovati in Europa e Nord America. Abbiamo anche notato che i ceppi virali trovati in Europa e Nord America derivano dal “ceppo” L- originario dell’Asia [23 ].
Caratterizzazione di hotspot geograficamente distinti nel tempo
Al fine di determinare l’aspetto di ogni mutazione, abbiamo analizzato nel tempo ogni genoma di ciascuna area geografica, classificandoli in base ai tempi di raccolta del campione, come indicato nel database GISAID. Secondo questa analisi, sono stati definiti 6 sottogruppi temporali, vale a dire dicembre 2019 (genomi di 5 pazienti), 1-15 gennaio 2020 (genomi di 15 pazienti), 16-31 gennaio 2020 (genomi di 52 pazienti), 1-15 Febbraio 2020 (genomi di 13 pazienti), 16-29 febbraio 2020 (genomi di 55 pazienti) e 1 °-13 marzo 2020 (genomi di 80 pazienti).
Il numero di mutazioni (normalizzate dalla popolazione presa in considerazione per ciascun periodo di tempo) aumenta nel tempo durante la diffusione virale fuori dall’Asia (vedi Fig. 2 ). Non sono state osservate mutazioni nei genomi asiatici analizzati a dicembre 2019. È interessante notare che un diverso modello di mutazioni è stato osservato in Europa tra gennaio e febbraio, quando è emersa una nuova mutazione, in posizione 14408 (raffigurata in rosso). Questa mutazione si trova nel gene RdRp. Sempre a partire da febbraio 2020, si osserva l’emergere di ulteriori nuove mutazioni (cioè 23403, 28881 e 2891 rispettivamente nero, blu elettrico, verde scuro). Nel tempo, abbiamo anche notato un aumento della frequenza della mutazione 3036 (arancione), già presente a metà gennaio (2,2%).
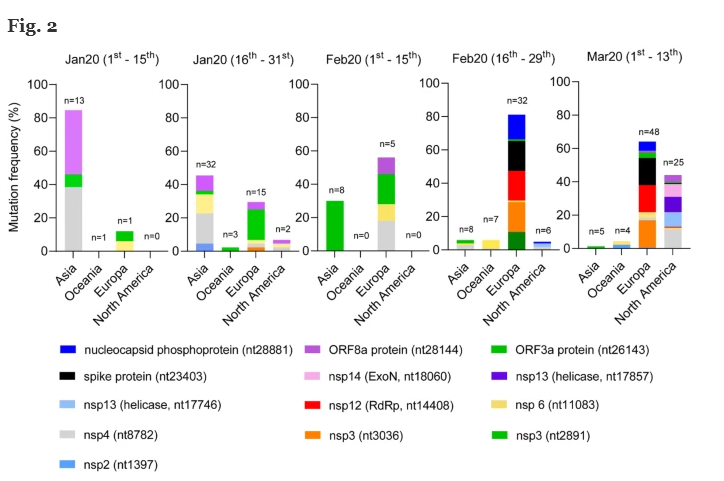
Inoltre, un diverso pattern di mutazioni hotspot è chiaramente distinguibile nei genomi virali rilevati nei pazienti nordamericani a partire da marzo 2020, quando è stato segnalato lo scoppio di casi positivi negli Stati Uniti e in Canada. In questo gruppo, sono state riportate tre nuove mutazioni (17746, 17857 e 18060, rispettivamente blu chiaro, viola e rosa chiaro). È interessante notare che i genomi virali presenti nei pazienti nordamericani portatori della mutazione RdRp (14%) non portano nessuna delle mutazioni specifiche europee.
Schema dei punti caldi delle mutazioni dopo il 9 febbraio 2020
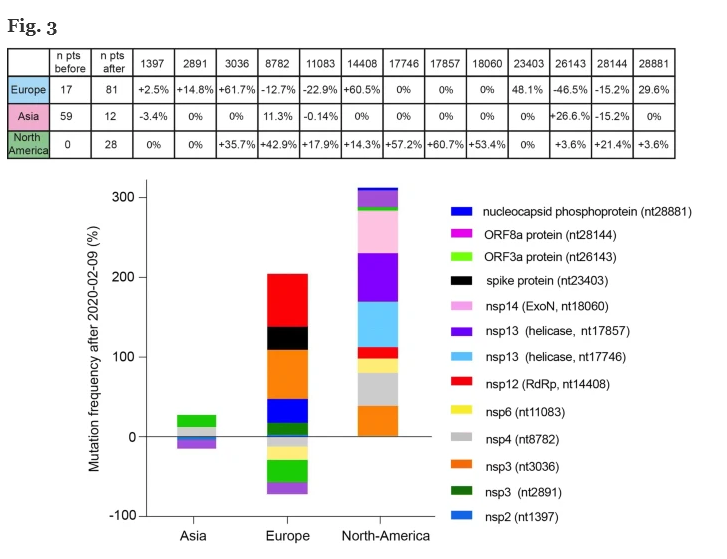
Data l’importanza di RdRp per la vitalità e la replicazione dei virus a RNA, è statisticamente meno probabile che si verifichino mutazioni in questo gene. Tuttavia, in alcuni casi, come nel poliovirus, sono stati riportati episodi di farmacoresistenza indotti da una mutazione puntiforme in RdRp [ 26 ]. Nel nostro database, la prima comparsa di una mutazione RdRp silenziosa (nt 14804) si è manifestata il 9 febbraio 2020 nel Regno Unito (Inghilterra), mentre per la prima volta si osserva una diversa mutazione RdRp (nt 14408, aminoacido da P a L) in Italia (Lombardia) il 20 febbraio 2020, quando è stato segnalato un drammatico aumento del numero di pazienti europei infetti dal sito web dell’OMS [ 27 ]. Abbiamo valutato l’aumento / diminuzione di ciascuna frequenza di mutazione prima e dopo il 9 febbraio 2020 nelle diverse aree geografiche (Fig. 3 ). In particolare, abbiamo osservato un forte aumento (+ 60,5%) dei genomi portatori della mutazione 14408 (che colpisce RdRp) in Europa, insieme ad un aumento dei genomi portatori della mutazione 3036 (+ 61,7%), della mutazione 23403 (48,1%) e la mutazione 28881 (+ 29,6%) (vedi tabella superiore Fig. 3 ).
Presenza simultanea della mutazione RdRp con altre mutazioni
Successivamente, abbiamo analizzato i genomi raccolti dopo il 9 febbraio 2020, quando per la prima volta nel database è stata segnalata la mutazione nel gene RdRp. Ai fini dell’analisi, abbiamo diviso i genomi in due gruppi: il gruppo 1 contiene genomi con mutazione in posizione 14408 (RdRp) (n = 53, 4 Nord America e 49 europei) e il gruppo 2 senza mutazione RdRp (n = 84) .
I genomi del gruppo 1 hanno mostrato un aumento del numero di mutazioni rispetto al gruppo 2. In particolare, il gruppo 1 mostra 6 genomi con due mutazioni (11,3%), 25 genomi con tre mutazioni (47,2%), 21 genomi con quattro mutazioni (39,6%) e 1 genoma con 5 mutazioni (1,9%). Nel gruppo 1, le mutazioni più segnalate sono quelle nelle posizioni 3036, 14408, 23403 e 28881. Per quanto riguarda i genomi nel gruppo 2, 20 non portano alcuna mutazione (23,8%), 25 genomi hanno una singola mutazione (29,8%), 19 i genomi hanno due mutazioni (22,6%), 6 genomi hanno tre mutazioni (7,1%), 9 genomi hanno quattro mutazioni (10,7%), 2 e 3 genomi hanno rispettivamente cinque e sei mutazioni (2,4% e 3,6%). Nel gruppo 2, le mutazioni più segnalate si trovano nelle posizioni 8782, 11083, 17746 e 17857.
La distribuzione tra i due gruppi in termini di numero di mutazioni è statisticamente rilevante (test Fisher-Exact, valore p <0,001). In particolare, i gruppi 1 e 2 sono significativamente differenti in termini di distribuzione dei genomi aventi 0, 1, 3 e 4 numeri di mutazioni (test di Fisher-Exact, p <0,001) (Fig. 4 ). Questa differenza, invece, è insignificante quando il numero di mutazioni è 2, 5 o 6.
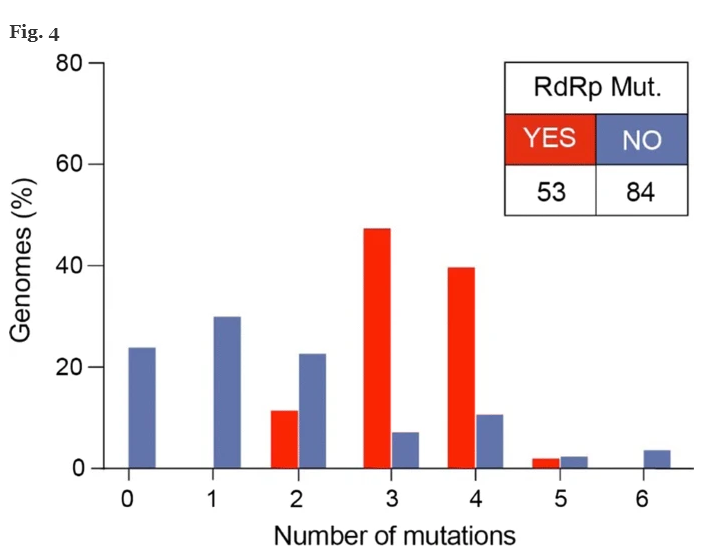
Abbiamo scoperto che i ceppi virali con mutazione RdRp hanno una mediana di 3 mutazioni puntiformi [range: 2-5], mentre i ceppi virali senza mutazione RdRp hanno una mediana di 1 mutazione [range: 0-3] (valore p <0,001, Mann –Test di Whitney). La diversa distribuzione tra i due gruppi rispetto al numero di mutazioni è statisticamente significativa (Fig. 4 ).
Abbiamo anche analizzato le mutazioni più frequenti rilevate: quelle nelle posizioni 3036, 23403 e 28881 (in Europa), e quelle nelle posizioni 17746, 17857 e 18060 (in Nord America). I genomi virali portatori di ciascuna di queste mutazioni sono stati confrontati con i genomi virali senza mutazioni, utilizzando il test di Mann-Whitney per l’analisi comparativa dei gruppi appaiati. I genomi portatori di mutazioni nelle posizioni 3036, 23403, 28881, 17746, 17857 e 18060 mostrano una mediana di 3-4 mutazioni (range [2: 5]), mentre i genomi portatori di nessuna di esse hanno una mediana di 1 o 2 mutazioni (range [ 0: 3], valore p <0,001, test di Mann-Whitney). Questa differenza è statisticamente significativa e implica che se una di queste mutazioni è presente, è più probabile che si verifichino altre mutazioni.
Studio di omologia della proteina RdRp mutante
Tra tutti i siti di mutazione analizzati, il mutante RdRp è particolarmente interessante dato che l’enzima è direttamente coinvolto nella replicazione virale e la sua fedeltà determina le capacità mutagene di SARS-CoV-2. A causa dell’elevata omologia tra RdRps di SARS-CoV e SARS-CoV-2, abbiamo allineato la sequenza di riferimento RdRp SARS-CoV-2 con la sequenza del sito catalitico riportata di SARS-CoV RdRp.
La sostituzione amminoacidica 323 (da P a L) (dovuta alla mutazione nucleotidica 14408) non rientra nel sito catalitico, in una regione che in SARS-CoV è segnalata come un dominio di interfaccia, una struttura superficiale ancora scarsamente caratterizzata, presumibilmente implicata nel interazione con altre proteine che possono regolare l’attività di RdRp [ 16 ]. A questo proposito, è noto che SARS-CoV RdRp forma un supercomplesso cilindrico cavo con nsp7 e nsp8, che conferiscono processività a RdRp [ 28]. Inoltre, il supercomplesso di replicazione interagisce con nsp14, un esonucleasi dotato della capacità di correzione di bozze tipica di Nidovirales. Questa attività è importante nel contesto del tasso di mutazione e per controllare la fedeltà nella replicazione dell’RNA. Tuttavia, i residui RdRp critici coinvolti in questa interazione devono ancora essere identificati e per questo motivo sono necessari ulteriori studi per valutare il possibile ruolo della mutazione 14408 riguardo alla fedeltà RdRp.
Discussione
Nel presente lavoro abbiamo confrontato il genoma di riferimento SARS-CoV-2 con quelli esportati dal database GISAID con l’obiettivo di ottenere importanti informazioni sulle mutazioni del virus, sulla loro occorrenza nel tempo e all’interno di diverse aree geografiche.
Abbiamo osservato che dopo febbraio 2020, quando sono stati segnalati i primi casi di SARS-CoV-2 trasmessi localmente dall’Asia, i genomi virali presentavano mutazioni puntiformi diverse, chiaramente distinguibili all’interno di diverse aree geografiche. Nel tempo, siamo stati in grado di identificare tre mutazioni ricorrenti in Europa (nelle posizioni 3036, 14408 e 23403) e altre 3 mutazioni diverse in Nord America (nelle posizioni 17746, 17857 e 18060). Finora, queste mutazioni non sono state rilevate in Asia. Il numero e la frequenza, nonché il valore mediano delle mutazioni puntiformi del virus registrate al di fuori dell’Asia, aumentano nel tempo.
Nel nostro studio, abbiamo scoperto che la mutazione RdRp, situata alla posizione 14408, che è presente nei genomi virali europei a partire dal 20 febbraio 2020, è associata a un numero maggiore di mutazioni puntiformi rispetto ai genomi virali dell’Asia. Dato che RdRp funziona in un macchinario complesso che include attività di correzione di bozze (in collaborazione con altri cofattori virali, come ExoN, nsp7 e nsp8), si è tentati di ipotizzare che questa mutazione abbia contribuito a compromettere la sua capacità di correzione di bozze. Un possibile meccanismo potrebbe comportare un piccolo cambiamento nella struttura RdRp, senza influire sulla sua attività catalitica, che potrebbe tuttavia alterare la sua capacità di legame con altri cofattori come ExoN, nsp7 o nsp8, alterando così il tasso di mutazione. Questo potrebbe spiegare l’aumento del numero di mutazioni che abbiamo osservato in Europa da febbraio 2020.Sono necessari ulteriori studi per determinare se le mutazioni a grappolo osservate provengono dallo stesso meccanismo molecolare. Sono inoltre necessari ulteriori studi per determinare se la mutazione in RdRp si traduca in una maggiore replicazione virale.
Alcuni inibitori della polimerasi [ 29 , 30 ] sono attualmente testati in studi clinici per colpire SARS-CoV-2 RdRp, tra cui Favipiravir [ 17 , 19 ], Galidesivir [ 18 ], Remdesivir [ 19 ], Ribavirin [ 20 ], Penciclovir [ 31 ], Galidesivir [ 32 ] e Ponatinib [ 33 ]. Inoltre, altri farmaci come Simeprevir (inibitore della proteasi dell’HCV approvato dalla FDA), così come Filibuvir e Tegobuvir (entrambi inibitori RdRp) [ 22], si prevede che leghino RdRp da studi di docking molecolare. In particolare, è stato identificato un putativo sito di attracco in una fessura idrofobica molto vicino al sito mutato 323 (da P a L), corrispondente alla mutazione 14408 identificata nel nostro studio [ 22 ]. Mutazioni naturali in RdRp possono potenzialmente portare a fenomeni di farmacoresistenza, come già osservato in precedenza [ 19 , 34 , 35]. In alternativa, potrebbe indurre una significativa diminuzione dell’affinità di legame del complesso farmaco-RdRp. Ciò potrebbe portare a una diversa efficacia dei trattamenti antivirali in cui è presente la mutazione 14408. Per questo motivo, data l’elevata frequenza di mutazione RdRp nella popolazione infetta, è importante caratterizzare l’impatto della mutazione 14408 sull’attività di RdRp e sulla sua suscettibilità ai farmaci antivirali.
Conclusioni
Abbiamo identificato nuovi hotspot di mutazione nelle sequenze del genoma SARS-CoV-2. È interessante notare che alcuni sono apparsi dopo febbraio 2020, solo nei pazienti europei. Tra questi hotspot, una mutazione nella posizione 14408 si trova all’interno della proteina RdRp ed è associata a un aumento del tasso di mutazione complessivo. Un’analisi in silico che confronta i domini funzionali annotati delle sequenze SARS-CoV e SARS-CoV-2, ha mostrato che questa particolare mutazione si verifica nel cosiddetto dominio dell’interfaccia RdRp, una struttura superficiale ancora poco caratterizzata, coinvolta nelle interazioni proteina-proteina [ 16]. Il ruolo per il dominio dell’interfaccia RdRp richiede ulteriori indagini, e in particolare l’effetto della mutazione in posizione 14408, la sua interazione con altri cofattori (come ExoN, nsp7 e nsp8), che possono influenzare la sua attività di correzione e potenzialmente alterare il suo tasso di mutazione. È anche essenziale capire se le mutazioni descritte potrebbero provocare l’emergere di fenotipi virali farmacoresistenti. I nostri dati possono aiutare lo sviluppo di strategie diagnostiche e terapeutiche e lo studio di potenziali meccanismi di resistenza ai farmaci.
Abbreviazioni
- RdRp:RNA polimerasi dipendente dall’RNA
- nsp:Proteina non strutturale
- SARS-CoV:Grave sindrome respiratoria acuta coronavirus
- SARS-CoV-2:Sindrome respiratoria acuta grave coronavirus 2 (COVID-19)
- MERS-CoV:Sindrome respiratoria mediorientale coronavirus
- ExoN:Esonucleasi
- CHI:Organizzazione mondiale della sanità
- WSM:Mercato del pesce di Wuhan