Alcuni organismi marini potrebbero contenere una cura per il melanoma,
il tipo più pericoloso di cancro della pelle.
Ascidians, or “sea squirts,” may hold the keys to a new anticancer compound. Credit: Wikimedia Commons
La cura per il melanoma – il tipo più pericoloso di tumore della pelle – potrebbe essere un composto derivato da un invertebrato marino che vive sul fondo dell’oceano? Lo pensano gli scienziati finanziati dalla National Science Foundation guidati da Alison Murray del Desert Research Institute di Reno, Nevada.
Stanno osservando il microbioma di un ascidico antartico chiamato Synoicum adareanum per comprendere meglio le possibilità di sviluppo di un farmaco specifico per il melanoma.
Gli ascidiani, o “schizzi di mare”, sono animali marini primitivi simili a sac che vivono attaccati ai fondali oceanici di tutto il mondo e si nutrono di plancton filtrando l’acqua di mare.
Il S. adareanum , che cresce in piccole colonie nelle acque circostanti l’Antartide, contiene un composto bioattivo chiamato “palmerolide A”, che ha promettenti proprietà anti-melanoma. I ricercatori ritengono che il composto sia prodotto da batteri che sono naturalmente associati a S. adareanum .
In un nuovo articolo pubblicato sulla rivista Marine Drugs , Murray e collaboratori dell’Università della Florida del Sud, il Los Alamos National Laboratory e l’Università di Nantes, Francia, presentano nuove scoperte sui batteri che compongono il microbioma di S. adareanum . Gli scienziati hanno misurato i livelli di palmerolide in campioni raccolti dall’Arcipelago Anvers Island dell’Antartide.
“Il nostro obiettivo a lungo termine è capire quale dei molti batteri in questa specie sta producendo palmerolide, ma per fare questo, c’è molto che dobbiamo imparare sul microbioma di S. adareanum “, ha detto Murray. “Il nostro nuovo studio descrive i numerosi progressi che abbiamo fatto verso tale obiettivo.”
Gli ecosistemi marini polari possiedono il potenziale per la biodiscovezione di composti bioattivi, in base alla loro diversità inutilizzata di macro e microrganismi. La caratterizzazione dei microbiomi polari bentonici associati agli invertebrati marini è limitata a pochi studi. Questo studio è stato motivato dal nostro interesse a comprendere meglio la struttura e la composizione del microbioma dell’ascide , Synoicum adareanum , in cui è stato isolato palmerolide A (PalA), un macrolido bioattivo con specificità contro il melanoma. PalA ha una somiglianza strutturale con un peptide-polichetide ibrido nonribosomico che ha somiglianze con i macrolidi prodotti microbicamente. Abbiamo condotto un’indagine spaziale per valutare sia i livelli di PalA sia la composizione del microbioma in S. adareanumin una regione della penisola antartica vicino all’isola di Anvers (64 ° 46 ′ S, 64 ° 03 ′ O). Il PalA era onnipresente e abbondante in una raccolta di 21 ascidiani (3 sottocampioni ciascuno) campionati da sette siti nell’arcipelago dell’isola di Anversa. La composizione del microbioma (varianti di sequenza del gene V3-V4 16S rRNA) di questi 63 campioni ha rivelato una suite di base di 21 varianti di sequenza di ampliconi batterici (ASV), 20 delle quali erano distinte dal batterioplancton regionale. L’analisi di co-occorrenza di ASV in tutti e 63 i campioni ha prodotto sottogruppi di taxa che potrebbero interagire biologicamente (sottosistemi interagenti) e, sebbene i livelli di PalA rilevati non siano stati trovati in correlazione con specifiche varianti di sequenza, i membri principali sembravano trovarsi in un ottimale preferito e intervallo di tolleranza dei livelli di PalA. Questi risultati,insieme a un’analisi del potenziale biosintetico dei relativi taxa di microbiomi, descrivono un microbioma centrale conservato ad alta latitudine con composizione unica e sostanziale promessa di biosintesi di prodotti naturali che probabilmente influenza l’ecologia dell’olobiont
I partner microbici degli invertebrati marini svolgono ruoli intrinseci nell’ambiente marino sia a livello individuale (sopravvivenza dell’ospite) sia a livello comunitario (distribuzione delle specie). Le relazioni ospite-microbo sono mediate da interazioni complesse che possono includere scambio di nutrienti, adattamento ambientale e produzione di metaboliti difensivi. Queste interazioni funzionali sono legate alla natura strutturale (diversità, biogeografia e stabilità) dell’ospite e del microbioma e alle interazioni ecologiche tra di loro. Studi su spugne, coralli e, in misura minore, ascidiani hanno rivelato forti tendenze nella specificità delle specie ospiti di invertebrati a particolari gruppi di batteri e archei. Questi studi hanno documentato uno strato sottostante di diversità (ad esempio, [ 1 , 2 , 3]) in cui l’habitat e la biogeografia sembrano avere forti influenze sulla struttura e sulla funzione del microbioma [ 4 , 5 , 6 ].
La stragrande maggioranza degli studi sul microbioma ospite è stata condotta a bassa e media latitudine dai siti costieri a quelli di acque profonde. Gli studi sul microbioma associato ad invertebrati marini bentonici ad alta latitudine sono attualmente limitati all’Antartico, dove è stata studiata solo la punta dell’iceberg per quanto riguarda le diverse associazioni ospite-microbo [ 7] e la comprensione ecologica è scarsa. Gli invertebrati marini antartici tendono ad avere un alto grado di endemicità a livello di specie, spesso presentano una distribuzione circumpolare e in molti casi hanno parenti più stretti associati alla fauna di acque profonde. Al momento non è noto se l’endemicità domini i microbiomi di invertebrati bentonici ad alta latitudine, né si comprende l’estensione della diversità all’interno e tra i diversi microbiomi associati all’ospite. Allo stesso modo, i rapporti sui microbiomi core (conservati all’interno di una specie ospite) all’interno delle specie di invertebrati antartici sono scarsi.
Finora i pochi studi sul microbioma associato all’ospite polare hanno documentato tendenze variabili nella specificità delle specie ospiti, con un numero generalmente basso di individui esaminati. Ad esempio, è stata segnalata una bassa specificità delle specie nelle composizioni di microbiomi di spugne tra diverse specie di Mycale sub-antartiche e delle Isole Shetland meridionali [ 8 ] che condividevano il 74% delle unità tassonomiche operative (OTU), che probabilmente rappresentavano un microbioma a nucleo incrociato Mycale . Al contrario, in cinque specie di spugne McMurdo Sound [ 9 ] sono stati osservati livelli elevati di specificità delle specie ospite-microbioma e sequenze core condivise all’interno di una specie . Lo stesso è stato trovato in diverse specie di spugne continentali antartiche [ 10 ]. Webster e Bourne [11 ] hanno anche trovato taxa batterici conservati dominati da microrganismi nella classe Gammaproteobacteria attraverso il corallo molle, Alcyonium antarcticum , campionati in tre siti in McMurdo Sound. Un altro cnidario, la nuova piattaforma di ghiaccio che scavava l’ anemone di mare Edswardsiella andrillae , conteneva un nuovo microbiota, sebbene la composizione attraverso un insieme limitato di individui fosse solo moderatamente conservata, in cui alcuni esemplari erano dominati da un OTU associato al phylum Tenericutes, e altri, un romanzo OTU nella classe Alphaproteobacteria [ 12 ]. Infine, un unico rappresentante dell’ascide antartica Synoicum adareanumha rivelato una limitata diversità nella sequenza genica di rRNA, inclusi rappresentanti di Actinobacteria, Bacteroidetes, Proteobacteria, Verrucomicrobia e TM7 phyla [ 13 ], sebbene la persistenza di questi taxa tra gli individui non sia stata studiata.
Alcyonium antarcticum (precedentemente, A. paessleri ) e Synoicum adareanum sono entrambi ricchi di metaboliti secondari. Il corallo molle A. antarcticum produce sesquiterpeni insoliti nel portare gruppi funzionali di estere nitrico [ 14 ], mentre l’ ascidiana, S. adareanum, produce una famiglia di polichetidi macrolidi, i palmerolidi, che hanno una potente attività contro il melanoma [ 15 ] ]. Il ruolo della comunità microbica nel contribuire a ospitare la chimica difensiva, le interazioni microbo-chimica e l’ottimizzazione della nicchia, nonché le interazioni microbo-microbo, non sono noti in questi ambienti ad alta latitudine.
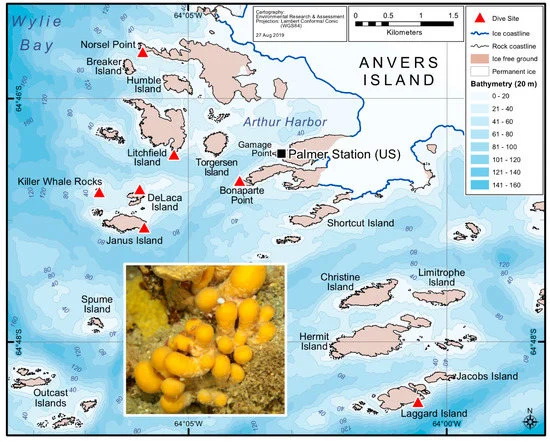
Qui, abbiamo progettato uno studio per indagare se un microbioma centrale persiste tra gli olobionti di S. adareanum che potrebbe informare la nostra comprensione delle origini di palmerolide. Abbiamo condotto un’indagine spaziale di S. adareanum in cui abbiamo studiato la quantificazione coordinata a livello di campione del principale metabolita secondario, palmerolide A (PalA), insieme alla diversità del microbioma associato all’ospite e alla struttura della comunità in tutto l’arcipelago dell’isola di Anvers (64 ° 46 ′ S, 64 ° 03 ′ O) sulla penisola antartica ( Figura 1 ). I risultati indicano una suite principale di microbi associati a S. adareanum contenente PalA , distinta dal batterioplancton, che porterà a test a valle dell’ipotesi che il produttore di PalA faccia parte del microbioma centrale
2.1. Variazione dei livelli di Holobiont PalA tra colonie ascidiane e siti di raccolta
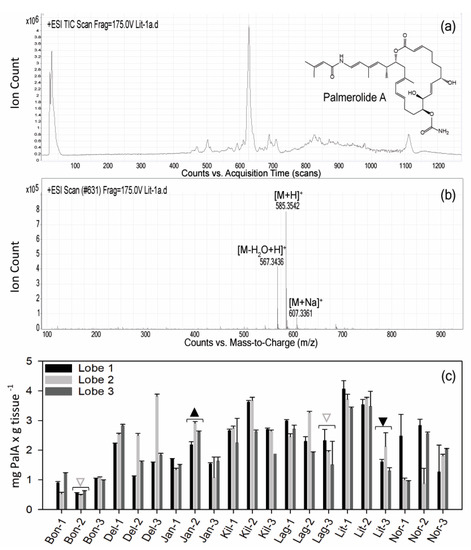
Le procedure tipiche per i campioni di chimica dei prodotti naturali utilizzano raccolte di campioni sfusi per l’estrazione chimica (~ 30 lobi ascidiani individuali per estrazione lipofila nel caso di S. adareanum ). Prima di questo studio, la variazione del contenuto di PalA a livello del lobo o della colonia individuale (inserto, Figura 1 ) era sconosciuta. Il nostro progetto di campionamento ha affrontato la variazione all’interno e tra le colonie in un determinato sito di campionamento, nonché tra le variazioni del sito. I siti erano limitati alla regione che era logisticamente accessibile dalla barca Zodiac nell’arcipelago di Anvers Island al largo della stazione di Palmer gestita dal Programma antartico degli Stati Uniti (USAP). Le colonie di S. adareanum sono state campionate attraverso sette siti di immersione ( Figura 1), in cui tre lobi per colonia a lobi multipli sono stati campionati da tre colonie per sito di immersione, per un totale di 63 lobi per il confronto PalA. Il PalA si distingue come il picco dominante in tutte le analisi LC-MS del diclorometano: frazione di metabolita solubile in metanolo di tutti i campioni analizzati (ad esempio, Figura 2 a, b). L’intervallo nei livelli di PalA variava con un ordine di grandezza di circa 0,49–4,06 mg PalA xg −1 peso a secco dell’ospite attraverso i 63 lobi esaminati. Il nostro disegno di studio ha rivelato lobo-a-lobo, livello di colonia all’interno del sito e alcune differenze da sito a sito nei livelli di PalA ( p <0,05) nell’arcipelago ( Figura 2 c e Figura S1). All’interno di una data colonia, la variazione da lobo a lobo era spesso elevata e significativamente diversa in 17 su 21 colonie esaminate. Differenze significative nei livelli di PalA tra le colonie sono state osservate anche in alcuni siti (Janus Island (Jan), Bonaparte Point (Bon), Laggard Island (Lag) e Litchfield Island (Lit); vedere la Figura 1 e la Figura 2c), in cui almeno una colonia aveva livelli significativamente diversi rispetto a un’altra o entrambe. Nonostante ciò, abbiamo trovato differenze tra alcuni dei siti. Vale a dire, Bon era significativamente inferiore rispetto a tutti e sei gli altri siti. Questo sito è il più vicino all’isola più grande, Anvers Island e Palmer Station. È stato anche riscontrato che campioni di siti di Killer Whale Rocks (Kil) e Lit hanno livelli di PalA significativamente più alti rispetto a Jan, Bon e Norsel Point (Nor), sebbene questi non sembrino avere un particolare schema spaziale o associazione con la profondità di raccolta dei campioni.
2.2. Caratterizzazione di batteri coltivati associati all’ospite
Dato il nostro interesse nell’identificare un microrganismo produttore di PalA [ 13 ], abbiamo eseguito uno sforzo di coltivazione con omogenato di S. adareanum su tre diverse formulazioni di mezzi marini a 10 ° C. Il sequenziamento del gene rRNA 16S ha rivelato sette isolati unici (di 16 portati in coltura pura) a un livello di identità di sequenza <99%. Tutti gli isolati tranne uno erano affiliati alla classe Gammaproteobacteria, inclusi cinque diversi generi comunemente isolati dagli ambienti marini ( Shewanella , Moritella , Photobacterium , dei quali nove erano altamente correlati). In molti casi, abbiamo caratterizzato i loro vicini vicini come noti psicofili marini, con molti da habitat polari ( Figura S2 Psychromonas e Pseudoalteromonas). L’eccezione a ciò è stata l’isolamento di una cultivar associata alla classe Alphaproteobacteria , Pseudovibrio sp. str. TunPSC04-5.I4, in cui i suoi due vicini più vicini erano isolati da una spugna marina temperata e un ascidico temperato (associato a una diversa famiglia di Ascidiacea). Questo risultato segna il primo Pseudovibrio sp. coltivato da alte latitudini. I risultati dello screening HPLC della biomassa da tutti i sedici isolati non hanno rivelato la presenza di PalA.
2.3. Synoicum Adareanum Microbiome (SaM)
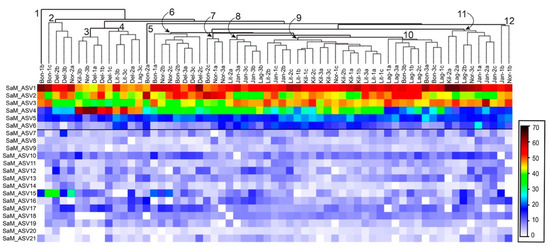
Per comprendere la natura della conservazione nella composizione del microbioma associato all’ospite di S. adareanum , abbiamo identificato la struttura e la diversità del microbioma (basato sulla regione V3 – V4 del gene 16S rRNA) con sezioni dei 63 campioni utilizzati per holobiont Determinazioni di PalA. Questo sforzo ha portato a 461 varianti di sequenza dell’amplicone del gene 16S rRNA (ASV) distribuite su 13 phyla batterici ( Tabella 1 , Tabella S1 ). La suite principale di microbi, definita come quella presente in> 80% dei campioni (indicato come Core80), comprendeva 21 ASV (sei dei quali erano presenti in tutti e 63 i campioni). Gli ASV Core80 rappresentavano la maggior parte degli iTag sequenziati (95% in media su tutti i 63 campioni), in cui i primi quattro ASV dominavano il set di sequenze (Figura 3 ). Gli ASV presenti nel 50% -79% dei campioni rappresentavano la categoria Dynamic50 e contenevano 14 ASV, che rappresentavano solo il 3,3% del set di dati. Gli ASV rimanenti sono caduti nella frazione variabile, che includeva 426 ASV, che rappresentano l’1,7% delle sequenze iTag, ma la maggior parte della ricchezza filogenetica ( Tabella 1). Sono state condotte analisi statistiche comparative con il set di campioni completo, che è stato ricampionato al numero più basso (9987) di iTag per campione. Questa procedura è stata limitata da un campione (Bon1b) che ha sottoperformato in termini di resa della sequenza iTag. Dopo l’eliminazione di questo campione dall’analisi, il set di 62 campioni aveva 19.003 iTag per campione con un totale di 493 ASV, le stesse 21 sequenze nel Core80 (con sette comuni a tutti i 62 campioni), le stesse 14 sequenze Dynamic50, e un totale di 458 ASV variabili.
La categoria di microbiomi Core80 includeva un grado relativamente alto di novità filogenetica, con quasi un terzo dei membri con identità di sequenza bassa (<95,0%) ai vicini più vicini. Allo stesso tempo, il resto degli ASV Core80 (14) corrispondeva a taxa marini per lo più incolti da mari polari o sedimenti, in molti casi con identità> 97%. Molte delle sequenze vicine più vicine sono originate da microbiomi di invertebrati marini. Gli ASV Core80 sono stati distribuiti su cinque phyla (Proteobatteri, Actinobacteria, Nitrospirae, Verrucomicrobia e Bacteroidetes; Tabella 1 ), in cui sette ASV altamente correlati sono stati associati al genere Gammaproteobacteria Microbulbifer e hanno dominato le sequenze ASV ( Figura S1). Altri due Gammaproteobacteria ASV nel Core80 sono stati affiliata alla Endozoicomonas genere ( SaM_ ASV7) e Nitrosospira ( SaM_ ASV13). C’erano diversi Alphaproteobacteria con i vicini più vicini che cadevano nei generi Pseudovibrio, Hoeflea, Sulfitobacter e Octadecabacter . Una sequenza legata a Rhodospirillales era solo lontanamente correlata a taxa noti, con un’identità di sequenza dell’86,6% al vicino più vicino. Un ASV correlato a Bprellovibrio Deltaproteobatteri faceva anche parte del microbioma Core80 ( SaM_ ASV9) che era anche unico, con un’identità di sequenza del 90,3% al suo vicino più vicino. Il core ASV affiliato con Actinobacteria (SaM_ ASV20) era correlato a una sequenza di Solirubrobacterales non coltivata. L’ASV11 corrispondeva più da vicino a una sequenza nella famiglia delle Nitrospirae dai sedimenti marini dell’Artico. Vi erano due sequenze associate alla Verrucomicrobium rappresentate in diverse famiglie ( Puniceicoccaceae , SaM_ASV14 e Opitutacae , SaM_ASV15). Infine, c’erano due ASV affiliati al phylum Bacteroidetes: uno legato alla deformazione polare, il Brumimicrobium glaciale (SaM_ASV19) e l’altro a una varietà di Lutibacter marina (SaM_ASV12).
Cinque ASV Dynamic50 sono stati affiliati al phylum marino di Bacteroidetes ( Cryomorphaceae e Flavobacteriacae ), oltre a sei ASV associati alla classe Gammaproteobacteria (incluse quattro sequenze aggiuntive correlate a Microbulbifer ). C’erano anche due phyla aggiuntivi, un ASV correlato a un isolato di Verrucomicrobium associato a spugna e un ASV correlato a Planktomycetes ( Tabella 1 , Figura S2 ). Molti di questi ASV erano strettamente correlati agli isolati dai sedimenti marini.
È interessante notare che cinque sequenze identificate dai precedenti sforzi di clonazione e sequenziamento con questo microbioma associato all’host ( Figura S2 ; [ 13 ]) corrispondevano a sequenze nei set di dati Core80 e Dynamic50. Confronti filogenetici hanno anche rivelato che gli isolati SaM erano distinti da Core80 e Dynamic50 ad eccezione di Pseudovibrio sp. TunPSC04-5.I4 isolato, che era presente sia nel Core80, sia nel clone e nello studio di sequenziamento.
Il raggruppamento gerarchico di ASV Core80 (basato sulla somiglianza di Bray-Curtis) tra tutti i 63 campioni non ha rivelato forti tendenze nei modelli specifici del sito o delle colonie ( Figura 3 ). Vi furono otto casi in cui due dei tre lobi si accoppiarono come vicini più vicini e quattro degli otto gruppi primari che includevano tre lobi derivati dalla stessa colonia. Esempio Bon1b raggruppato a parte tutti. In alcuni casi, i cluster potrebbero essere attribuiti a ASV specifici. Ad esempio, il cluster 2 ( Figura 3 ) presentava i livelli relativi più alti di SaM_ASV15 (una sequenza affiliata alla famiglia delle Opitutaceae ), mentre il cluster 3 ( Figura 3 ) presentava i livelli relativi più alti di ASV4 (affiliato con Microbulbifer spp.).
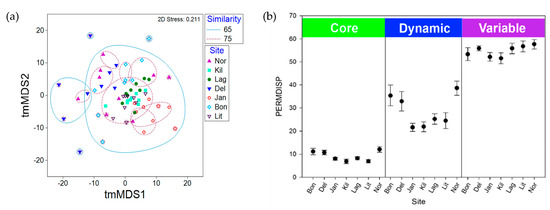
Complessivamente, le strutture comunitarie nei microbiomi di S. adareanum nei 63 lobi esaminati presentavano un alto grado di somiglianza. I confronti di somiglianza a coppie di Bray-Curtis tra lobi e colonie all’interno di ciascun sito erano in ogni caso superiori al 54%. Quando si confrontano le medie dei valori di somiglianza a coppie all’interno e tra le colonie, tutti i siti, ad eccezione di Lag, avevano valori di somiglianza più elevati all’interno dei lobi della stessa colonia (che vanno dal 69,9% all’82,2%) rispetto alle colonie all’interno di un sito (66,5% –81,0 %; Figura S3 ), anche se le differenze erano piccole e solo Bon e Jan erano significativamente diverse ( p <0,05). Abbiamo effettuato un’analisi tmMDS bidimensionale basata sulla somiglianza di Bray-Curtis per studiare la struttura del microbioma tra i siti (Figura 4 a). I microbiomi campionati a Kil e Lit avevano il più alto grado complessivo di clustering (> 75% di somiglianza) mentre i campioni di Kil, Lag, Lit e Jan erano tutti raggruppati a un livello del 65%. I microbiomi di DeLaca (Del) erano i più diversi, supportati dall’analisi SIMPER, in cui due degli ASV più abbondanti nel Core80 erano inferiori alla media tra gli altri siti (SaM_ASV1 e 3) mentre altri nel Core80 (SaM_ASV4 , 15 e 17) erano superiori alla media tra i siti. Abbiamo anche eseguito un tmMDS 2D sulle frazioni SaM (Core80, Dynamic50, Variable) con e senza iterazioni permutazionali, che ha mostrato tendenze simili sebbene il partizionamento delle strutture della comunità tra i siti fosse più evidente con le permutazioni ( Figura S4). I modelli di clustering basati sul sito si sono spostati in una certa misura nelle diverse frazioni SaM. Solo per il Core80, i campioni di Jan si sono raggruppati separatamente dagli altri. Per Dynamic50, sia Jan che Kil erano valori anomali. Infine, per la frazione variabile, i campioni Kil e Del si sono raggruppati separatamente dagli altri siti. La frazione variabile era più omogenea, oscurando qualsiasi variabilità da sito a sito, mentre il nucleo mostrava nuvole di dati più strette che mostravano un modesto livello di dispersione.
Un’analisi PERMANOVA ha studiato i driver della variabilità nella struttura della comunità, in cui abbiamo testato il ruolo della variazione ambientale a livello di colonia, a livello di sito e stocastica. Nel valutare l’intera comunità (tutti i siti e ASV), le differenze da sito a sito hanno spiegato il 25% della variabilità nel microbioma ( Tabella S2 ). Le differenze colonia-colonia hanno spiegato il 28%, mentre il rimanente 47% della variabilità era inspiegabile ed è probabilmente attribuito alla variazione ambientale stocastica. Quando le frazioni SaM sono state analizzate separatamente, la differenza più significativa ( p <0,05) era nella frazione variabile del microbioma, in cui le differenze da sito a sito spiegavano solo il 19,2% della variazione e il livello residuo (stocastico) aumentava al 58,4% ( tabella S2). Al contrario, PERMDISP ( Figura 4 b), che rappresenta la dispersione attorno al centroide calcolato per ciascun sito (una misura della β-diversità), ha rivelato differenze nelle strutture della comunità in ciascuna frazione SaM tra i siti, nonché differenze tra le frazioni del microbioma. Il Core80 aveva un basso livello di dispersione (media PERMDISP di 9,1, intervallo: 6,8-12,1), rispetto al Dynamic50 (media 28,6, intervallo: 24,5-38,6), che erano moderatamente dispersi attorno al centroide ed erano più variabili con differenze tra i siti più apparenti; Bon, Del e Nor avevano valori più alti rispetto agli altri quattro siti. La frazione variabile aveva valori PERMDISP elevati (media 54,7, intervallo: 51,6-57,6), che rappresentavano elevate differenze nella diversità β, in cui i valori erano relativamente vicini tra i sette siti.
2.4. Co-occorrenze di ASV e relazione con il PalA
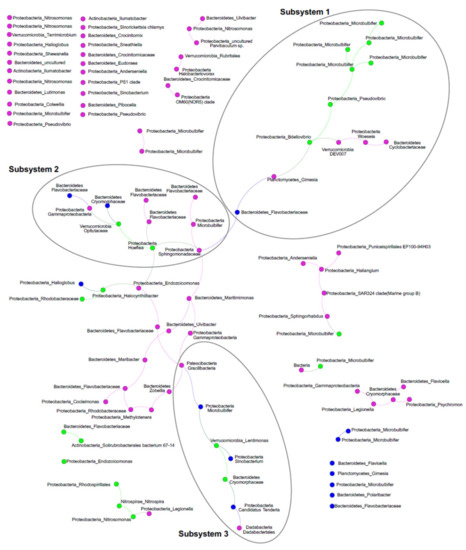
Per studiare ulteriormente l’ecologia ascidiana-microbioma, abbiamo eseguito un’analisi di rete dell’olobiont di ascide con particolare enfasi sulla co-occorrenza di ASV ed esplorato la relazione tra ASV e PalA usando misure dirette e indirette. La rete di ricorrenza rappresentava un grafico rado con 102 nodi e 64 spigoli (grado medio 1,255; diametro 15; lunghezza media del percorso 5,81) che associava gli ASV da frazioni SaM simili (coefficiente di assortimento di 0,41), a conferma della solidità di cui sopra. A seguito dell’ispezione della rete di ricorrenza, un componente connesso dominante conteneva il 42,1% degli ASV, in cui abbiamo identificato tre moduli altamente connessi (tramite l’applicazione di un’analisi di rete indipendente WGCNA, soglia morbida = 9, qui indicati come sottosistemi; Figura 5). La rete conteneva anche diversi sistemi più piccoli (2-6 nodi) e 30 singleton, solo uno dei quali era nel Core80 (classe Gammaproteobacteria, Endozoicomonas- affiliata). I tre sottosistemi includevano ASV delle frazioni Core80, Dynamic50 e Variable SaM, ma erano distribuiti in modo non uniforme. Principalmente guidato dalle frazioni Core80 SaM, il sottosistema 1 ospitava la maggior parte degli ASV altamente rappresentati, compresi gli ASV correlati a Microbulbifer e l’ ASV Pseudovibrio . Sottosistema 2, interconnessione sottosistemi 1 e 3, è in gran parte guidato da ASV frazione variabile e comprende inferiore rispetto abbondante Core80 Hoeflea e Opitutaceae ASV, così come diverse Bacteroidetestaxa. Infine, il sottosistema 3 era più piccolo. Comprendeva diversi taxa diversi dominati dalla frazione Dynamic50, tra cui Lentimonas e ASVs affiliati a Cryomorphaceae dal Core80. Il ruolo complessivo degli ASV variabili nella rete è sorprendente, in quanto sembrano essere nodi critici che collegano i sottosistemi attraverso 13 spigoli che si trovano tra i sottosistemi 2 e 3. Un altro componente forse forse più piccolo connesso si trova nella parte inferiore sinistra del grafico, in quali tre ASV Core80, l’ammoniaca chemoautotrofica e gli ossidanti nitriti, Nitrosomonas e Nitrospira e un ASV insolito correlato a Rhodospirillales, erano collegati con un ASV variabile.
Per rispondere alla domanda del primo ordine sulla presenza di una relazione tra i livelli di concentrazione di PalA rilevati nell’analisi LC-MS e le occorrenze semiquantitative di AS adareanumASV, abbiamo eseguito tre analisi complementari: analisi di correlazione, analisi ponderata delle reti di ricorrenza (WGCNA) e optima di nicchia robusta con concentrazione di PalA come variabile. Le correlazioni di Pearson tra le occorrenze di ASV e le concentrazioni di PalA variavano da -0,33 a 0,33 al massimo (24 delle quali erano significative, ≤ 0,05; sebbene nessuna di quelle con una relazione significativa facesse parte del microbioma centrale e fosse presente in <50% del campioni con la più alta occorrenza di 24 sequenze), suggerendo una scarsa relazione a livello lordo di PalA normalizzato a peso secco e abbondanza relativa di ASV. Ciò ha indicato che l’abbondanza relativa di ASV non è un buon predittore di PalA. Un risultato WGCNA complementare non ha mostrato alcuna associazione significativa tra topologia di co-occorrenza del sottosistema e PalA (correlazioni = −0,23; 0,033; −0.17 per i sottosistemi 1, 2 e 3 rispettivamente), indicando la mancanza di relazione tra la struttura della comunità microbica e il PalA. Infine, è stato esplorato un altro aspetto della relazione tra occorrenza di ASV e livelli di PalA utilizzando il metodo ottimale robusto, che stima l’intervallo ecologico ottimale e la tolleranza per gli ASV sul PalA. In questo caso, abbiamo calcolato l’ottimale di nicchia PalA per ciascun ASV e li abbiamo classificati in base alla mediana (abbiamo calcolato l’ottimale di nicchia PalA per ciascun ASV e li abbiamo classificati in base alla mediana (abbiamo calcolato l’ottimale di nicchia PalA per ciascun ASV e li abbiamo classificati in base alla mediana (Figura S5 ). Gli ASV Core80 hanno mostrato una gamma di nicchie PalA coerente. Inoltre, i valori medi di optima degli ASV Dynamic50 si trovano, per la maggior parte, in optima PalA inferiore o superiore rispetto al Core80, con una sostanziale sovrapposizione di nicchia tra Core80. Gli ASV variabili si trovano collettivamente agli estremi inferiore e superiore dell’intervallo ottimale e di tolleranza. Complessivamente, queste analisi complementari sostenevano di non considerare un effetto individuale o comunitario sul PalA, ma piuttosto l’acclimatazione agli alti livelli di PalA osservati ( Figura 2 c) che probabilmente si basano su controlli metabolici o ambientali sconosciuti.
2.5. Raccolta delle colture: confronto tra microbioma e batterioplancton
Per stabilire se la composizione delle cultivar batteriche associate al Synoicum adareanum fosse presente anche nel SaM o nel batterioplancton a vita libera (frazione <2,5 μm), abbiamo considerato la sovrapposizione tra le sequenze del gene dell’isolato 16S rRNA e queste altre due set di dati. La rappresentazione degli isolati nel set di dati ASV del gene 16S rRNA è stata stimata confrontando i due set di dati di sequenza, sebbene diversi campioni di ascidiana siano stati utilizzati per la raccolta di colture e il sondaggio di S. adareanum . Tranne Pseudovibriosp. TunPSC04-5I4 che era presente nel Core80 con una corrispondenza della sequenza del 100%, altri due isolati avevano anche corrispondenze del 100% alle sequenze nella variabile SaM (BONS.1.10.24 e BOMB.9.10.19). Altre tre sequenze isolate di rRNA 16S (BOMB.3.2.14, BOMB.9.10.16, BOMB.9.10.21) sono state trovate per abbinare le sequenze nel microbioma variabile a un livello del 97% o superiore. Solo le sequenze di isolati correlati a Pseudoalteromonas e le sequenze di isolati correlati a Shewanella non sono state rilevate con parenti con un livello di identità di almeno il 97% rispetto agli ASV SaM; che potrebbe essere spiegato con il sottocampionamento. La composizione del batterioplancton era dominata dagli ASV di Gammaproteobacteria (47,35% di tutti gli ASV; Tabella S3), in cui sei degli isolati (BOMB.9.10.21, BONS.1.10.24, BOMB.9.10.16, BOMB.3.2.20, BOMB.3.2.14, BONSW.4.10.32) corrispondevano a sequenze nel batterioplancton set di dati con identità> 99,2%; anche a un livello di identità di sequenza del 95%, i restanti dieci isolati non corrispondevano a sequenze nel plancton, incluso lo Pseudovibrio sp. str. TunPSC04-5I4 isolato.
2.6. Microbioma: confronti del batterioplancton
Sebbene a livello tassonomico elevato i proteobatteri e i batterioideti phyla dominassero il microbioma e il batterioplancton ( Tabella S3 ), le proporzioni relative variavano e i taxa rappresentati erano piuttosto diversi. L’appartenenza tra SaM e il set di dati del batterioplancton indicava una sovrapposizione di basso livello al 100% di identità, con 39 dei 604 ASV perfettamente corrispondenti. Con un’identità di sequenza ASV al 100%, i risultati indicano che un singolo ASV Core80 SaM era un abbinamento perfetto con il set di dati del batterioplancton: il Microbulbifersequenza associata che è la più abbondante in tutti i 63 set di dati SaM. È interessante notare che questa sequenza è stata identificata in un solo campione di batterioplancton (IPY-225-9) a bassa occorrenza (14 di> 1,18 milioni di tag distribuiti su 604 ASV). Esistevano tre (di 14) ASV Dynamic50 che corrispondevano perfettamente agli ASV di batterioplancton. Questi erano affiliati con una Flavobacteraceae ASV della famiglia dei Bacteroidetes scarsamente classificata (77% dei campioni SaM), un Sinobacterium associato a Gammaproteobatteri (73% dei campioni SaM) e un secondo ASV associato a Gammaproteobacteria associato a CandidatusTenderia (71% dei campioni SaM). I restanti 35 ASV con corrispondenza perfetta tra i due set di dati sono stati classificati come parte del microbioma variabile. Queste sequenze sono state suddivise in quattro phyla e nove classi, 13 delle quali sono state ben distribuite su campioni di set di dati sul batterioplancton (> 50%).
A un livello di identità della sequenza ASV del 97%, c’erano tre ulteriori corrispondenze tra il set di dati del batterioplancton e il Core80. Questi includevano sequenze correlate a Hoeflea e Halocynthibacter correlate a Alphaproteobacteria e una sequenza correlata a Nitrospira . C’erano anche altri due ASV correlati a Dynamic50: entrambi erano correlati a Flavobacteriaceae non classificati . Il resto (79 ASV) delle partite con> 97% erano affiliati alla frazione variabile del microbioma.
Questo studio riporta la nostra crescente comprensione della composizione del microbioma di S. adareanum contenente PalA . Per migliorare la nostra comprensione dell’ecologia dell’ascidiana contenente PalA, S. adareanum , abbiamo studiato il microbioma della colonia di ascide e i livelli di chimica del PalA a livello di lobo individuale, e confrontato il microbioma di ascide con il plancton. Questo confronto ci ha permesso di rispondere a domande riguardanti la variabilità del contenuto di PalA e se si verifica un microbioma centrale conservato attraverso questi ascidici antartici contenenti PalA, supportando così la logica che se un produttore microbico sintetizza il PalA, l’organismo produttore dovrebbe essere presente in tutto il PalA contenente S. adareanumcampioni. Prima di questo studio, tuttavia, non avevamo dati quantitativi a livello del singolo lobo ascidiano che forma le colonie peduncolate di S. adareanum ( Figura 1 ). Questa discussione si concentra sul microbioma centrale, quindi esamina più in dettaglio le distribuzioni secondarie di metaboliti sul microbioma e in altri invertebrati marini, nonché il potenziale biosintetico dell’appartenenza al nucleo, e si conclude con le informazioni acquisite dal nostro sforzo iniziale di coltivazione.
3.1. Microbioma core
La specificità del microbioma ascidico (ospite) è un’area attiva di ricerca. Rispetto alle spugne e ai coralli, ad esempio, i microbiomi di ascide sono stati meno caratterizzati. Per ottenere una prospettiva ampia sulla composizione del microbioma, abbiamo anche usato approcci indipendenti dalla coltivazione. Abbiamo scoperto che l’ ascidia antartica S. adareanum ha un microbioma centrale persistente nell’arcipelago dell’isola di Anvers che è distinto dal plancton. Questa differenza tra i microrganismi associati all’ospite di ascide e il batterioplancton sembra essere un’osservazione coerente attraverso l’oceano globale (ad esempio, [ 16 , 17 , 18 , 19]). Il Core80 è composto da ASV che dominano numericamente la comunità, così come quelli che rappresentano solo una frazione di una percentuale delle sequenze esaminate. Sebbene le simbiosi di ascide non siano state ancora sistematicamente studiate in Antartide, i microbiomi di ascide a latitudine inferiore meglio studiati forniscono diversi esempi per il confronto. La tendenza generale degli studi sul microbioma di ascidia fino ad oggi suggerisce che esiste un alto grado di specificità sia a livello geografico che a livello di specie ospite della composizione del microbioma (ad esempio, [ 16 , 17 , 20 ]). Lo stesso sembra essere vero per S. adareanum. Sebbene questo studio fosse limitato a una piccola regione geografica, abbiamo identificato un nucleo conservato di 21 tipi di sequenza genica del rRNA 16S su 63 singoli lobi peduncolati studiati. Attribuiamo il rilevamento di questo elevato grado di membri persistenti in parte all’omogeneizzazione, all’estrazione e alla pipeline metodologica applicate. L’analisi del microbioma è sensibile alla profondità di sequenziamento, alla scelta dei parametri di qualità e alle differenze algoritmiche nelle condotte di elaborazione dei dati (varianti della sequenza degli ampliconi rispetto alle unità tassonomiche operative derivate dal cluster), che possono influire sul confronto diretto tra gli studi. Lungo queste linee, i numerosi Microbulbifer altamente correlatiGli ASV sarebbero caduti in un singolo OTU (identità di sequenza del 97%), risultando in un nucleo con 14 membri. A parte queste limitazioni, i nostri risultati sono in linea con molti altri studi sul microbioma di ascide da latitudini più basse in termini di dimensioni relative dell’appartenenza del nucleo (dove le definizioni del nucleo variano in una certa misura tra gli studi). Ad esempio, è stato riferito che Styela plicata , un’ascida solitaria, aveva un nucleo di appartenenza da 10 [ 21 ] a 16 OTU [ 22 ]. Altri ascidiani solitari, tra cui Herdmania momus, avevano un nucleo di 17 OTU [ 21 ], mentre due specie Ciona variavano da 8 a 9 OTU [ 23 ]. Ascidiani coloniali temperati Botryliodes leachi eBotryllus schlosseri variava da 10 a 11 membri nei loro microbiomi core [ 20 ]. Inoltre, un ampio sondaggio su 10 diversi microbiomi di ascide (che rappresentano sia forme solitarie che coloniali) condotto sulla Grande Barriera Corallina ha riportato un numero di membri chiave compreso tra 2 e 35 OTU [ 16 ], mentre il numero di individui intervistati in ciascun caso era solo di 2– 3. Si noti che alcuni altri studi hanno riportato un numero molto più elevato di OTU condivise che vanno da 93 a 238 [ 18 , 19 ]; la scala del sequenziamento era più alta in questi studi successivi. Inoltre, come altri hanno riferito [ 24], l’appartenenza a questi microbiomi ascidici core è distinta e, nel caso del SaM, la diversità dei microbiomi core sembra essere unica a livello di ASV, sebbene diversi taxa siano in comune con altri microbi associati all’ascidiana a livello di genere, incluso Microbulbifer associato a Cystodytes sp. [ 25 ], Pseudovibrio con Polycitor proliferus [ 26 ] e un clade specifico Endozoicomonas sono stati identificati in un sondaggio di un numero di ascidiani [ 27 ].
Le capacità metaboliche previste dei taxa Core80 suggeriscono che l’eterotrofia aerobica (respirazione aerobica – il carbonio organico è la fonte di carbonio ed energia), la microaerofilia (crescita in condizioni di basso ossigeno) e la chemioautotrofia (la fissazione della CO 2 fornisce carbonio e sostanze chimiche ridotte forniscono energia, ad esempio NH 4 + e NO 2 – ) sono temi tra i Core80, in cui gli ASV più abbondanti sono i degradatori del carbonio ad alto peso molecolare. Il genere Microbulbifer ha membri noti per degradare la cellulosa [ 28 ] e, forse per coincidenza, gli ascidiani sono gli unici invertebrati conosciuti in grado di biosintesi della cellulosa nell’ambiente marino (ad esempio, [ 29 , 30]). Da ciò, potremmo ipotizzare che i ceppi di Microbulbifer associati a S. adareanum potrebbero occupare una relazione commensale, se non in qualche modo antagonista [ 31 ]. A sostegno di questa possibilità è il fatto che l’unica sequenza sovrapposta tra il Core80 e il batterioplancton era una sequenza microbulbifera , che era una rara sequenza nel plancton, suggerendo che potrebbe essere un membro opportunistico del microbioma di S. adareanum . Inoltre, è stato scoperto che gli isolati a vita libera e associati alla spugna del genere Microbulbifer producono composti bioattivi tra cui pelagiomicine [ 32 ] e parabeni [ 33], rispettivamente. Questa osservazione, perlomeno, suggerisce che i Microbulbifer (s) sono / sono probabilmente ben adattati al loro ospite ascidico e potrebbero essere considerati un potenziale organismo produttore di PalA.
NH 4 + -oxidizing Nitrosopumilis -affiliated Thaumarchaeota sono stati comunemente rilevati in microbiomes ascidie [ 16 , 21 , 22 , 34 ], che contrasti filogeneticamente, ma non in termini di funzione biogeochimico, con NH 4 + -oxidizing Nitrosomonas ASV che era parte del core SaM. La nicchia, tuttavia, è stata segnalata come diversa per gli ossidanti NH 4 + arcaici e batterici , in cui gli archaea tendono a trovarsi nei sistemi oligotrofici, mentre i batteri (ad esempio Nitrosomonas ) possono tollerare alti livelli di ammoniaca disciolta (rivista di [35 ]). Questo risultato potrebbe riflettere sia l’ambiente, sia i livelli di ammoniaca in situ del tessuto di S. adareanum dove potrebbe accumularsi. Numerosi studi hanno riportato gli alti livelli di ammoniaca ossidante Thaumarchaeota nelle acque costiere dell’arcipelago dell’isola di Anvers che sono numerosi solo in inverno fino alle prime acque di sorgente [ 36 , 37 , 38 ]. Il nostro studio è stato condotto con campioni raccolti in autunno, quando i Thaumarchaeota ossidanti l’ammoniaca non sono abbondanti nell’acqua di mare costiera [ 36 ], sostenendo i confronti tra il SaM e il batterioplancton raccolti in entrambi i periodi di fine estate e inverno.
Allo stesso modo, sebbene non abbiamo condotto intenzionalmente uno studio temporale, i dati dei campioni raccolti nel 2007 e nel 2011 sembrano suggerire che alcuni microrganismi core sono stabili nel tempo. Nel 2011 abbiamo trovato diversi ASV che corrispondevano a sequenze clonate (al 100% di identità di sequenza) da campioni raccolti nel 2007 [ 13 ]. La stabilità del microbioma ascidico nel tempo è stata riportata in alcuni studi [ 17 , 24 , 34 ]. Sarebbe interessante studiare la persistenza dell’appartenenza di base nel ciclo annuale (e fornire prove convincenti per relazioni stabili) in questo ambiente ad alta latitudine, dove luce, produzione di carbonio e copertura del ghiaccio marino sono molto variabili.
L’analisi di ricorrenza ha indicato tre sottosistemi di ASV che si verificano contemporaneamente in S. adareanum . Una piccola rete laterale includeva i due taxa coinvolti nel processo di nitrificazione in 2 fasi, tra cui l’ ASV Nitrosomonas sopra menzionato e un ASV Nitrospira . Anche se al momento, le basi funzionali del sistema ospite-microbico non sono state studiate, le relazioni di ricorrenza forniscono foraggio per il test di ipotesi in futuro. Una rete di interazione che merita di essere menzionata qui sono gli ASV nel sottosistema 1, che ospitano numerosi ASV Micro80 Microbulbifer Core, e gli ASV Pseudovibrio sono collegati a un ASV Bdellovibrio che è anche membro del Core80. Membri delIl genere bdellovibrio è un predatore batterico obbligato [ 39 ] che penetra nella membrana esterna e nella parete cellulare della preda. La posizione di collegamento nel sottosistema è convincente nel senso che il Bdellovibriopotrebbe potenzialmente controllare l’abbondanza dei membri connessi della rete. Infine, le posizioni di un paio di Dynamic e diversi ASV variabili nella rete, come collegamenti tra i sottosistemi, erano inaspettate. Le posizioni centrali di questi ASV suggeriscono che potrebbero non essere semplicemente membri stocastici del microbioma; che potrebbero svolgere ruoli opportunistici, adattivi o ecologici nella funzionalità dei sottosistemi di microbiomi che potrebbero potenzialmente partecipare a diversi aspetti del sistema holobiont in particolare, promuovendo il passaggio tra le diverse modalità ecologiche supportate da diversi sottosistemi. Tali ruoli sono stati proposti per i membri dinamici del microbioma Styela plicata [ 21 ].
Lo sforzo di coltivazione è riuscito a isolare un ceppo Pseudovibrio che è un membro cruciale del Core80. Inoltre, sono stati coltivati molti altri ceppi affiliati a Gammaproteobacteria che corrispondevano a sequenze nel SaM variabile e nel batterioplancton. Tuttavia, la diversità coltivata utilizzando gli approcci applicati qui ha generato una raccolta di diversità limitata. È probabile che ulteriori tipi di media e strategie di isolamento possano tradursi in un’ulteriore diversità coltivata in quanto vi sono un certo numero di taxa con stili di vita eterotrofi aerobici nel Core80 che sono stati portati nella cultura pura (ad esempio, Microbulbifer , Hoeflea ). Una delle sfide che abbiamo riscontrato nell’uso dei terreni ricchi di nutrienti è stata la crescita eccessiva di lastre, anche a 10 ° C.
3.2. Distribuzioni secondarie di metaboliti e bioaccumulo nel biota marino
Sebbene i risultati del sondaggio sull’ascidia spaziale dell’arcipelago non abbiano supportato una relazione diretta tra i livelli di PalA e la relativa abbondanza di ASV di microbiomi, i risultati dell’analisi di nicchia di PalA suggeriscono che gli ASV di Core80 si verificano in un intervallo ottimale e di tolleranza preferito dei livelli di PalA. La mancanza di specifici schemi ASV-PalA potrebbe non essere del tutto sorprendente, poiché i metaboliti secondari derivano da una complessa combinazione di reazioni metaboliche che richiedono un perfezionamento delle condizioni ambientali e un’ulteriore modellizzazione metabolica a fini di comprensione. Inoltre, è stato scoperto che questi metaboliti si accumulano nei tessuti in diversi invertebrati marini.La teoria della difesa ottimale può essere applicata agli invertebrati marini e riflette l’ipotesi che i metaboliti secondari siano distribuiti in tessuti specifici in base all’esposizione e alla suscettibilità anatomica alla predazione [39 ]. Ad esempio, i nudibranchi sequestrano i composti tossici, che sono stati biosintesi dal gasteropode o acquisiti dalle loro prede. Le tossine sono concentrate nello spazio anatomico dei loro mantelli, la parte più vulnerabile dei loro corpi molli ed esposti [ 40 , 41 , 42 ]. È noto anche il bioaccumulo di metaboliti secondari negli invertebrati con meno differenziazione anatomica. Nel phylum Porifera sono stati studiati diversi tipi e strati cellulari per determinare le differenze spaziali e anatomiche nelle concentrazioni secondarie di metaboliti [ 43 , 44 , 45 ]. È stato scoperto che i composti sono concentrati spazialmente sulla superficie (ad esempio, [ 46]) o parti apicali della spugna [ 47 ] in alcuni casi. Le spugne possono essere in grado di bioaccumulare in modo differenziato i metaboliti citotossici secondari in base a tessuti più sensibili alla predazione [ 48 ]. Le indagini sulla distribuzione dei metaboliti specifiche dell’ascidiana sono meno ben documentate; tuttavia, ci sono anche prove del bioaccumulo del metabolita citotossico secondario dell’ascidia. I patellazoli, macrolidi marini della rotula del Lissoclinum di ascide , si bioaccumulano nei tessuti di ascide a concentrazioni fino a sette ordini di grandezza superiori alla loro dose citotossica nelle linee cellulari dei mammiferi [ 49 , 50]. Inoltre, vi sono altri casi in cui il bioaccumulo nei tessuti ospiti dell’ascidiana suggerisce una cooperazione metabolica tra produttore e ospite, nonché traslocazione del composto dal produttore all’ospite [ 15 , 51 , 52 ]. Sebbene i livelli di PalA siano stati normalizzati a grammi di peso del lobo secco, la localizzazione spaziale specifica del tessuto è un fattore potenzialmente confondente nelle analisi statistiche che studiano la relazione ASV: PalA.
3.3. Il potenziale biosintetico del nucleo
Abbiamo studiato il potenziale biosintetico del prodotto naturale dei nove generi associati a 15 dei 21 ASV Core80 usando antiSMASH ( Tabella 2 , Tabella S4 ). Da ciò sembra che tutti i generi avessero almeno un parente a livello di genere con capacità biosintetica per la biosintesi del polichetide o del peptide nonribosomiale o di entrambi. Anche se il numero di genomi disponibili per il sondaggio era altamente irregolare, esiste una disparità di capacità biosintetica tra i generi analizzati, quindi sembra che Pseudovibrio , Nitrosomonas , Microbulbifer , Nitrospira abbiano le maggiori capacità (in questo ordine). Allo stesso modo, Microbulbifer , Pseudovibrio, Hoeflea e Opitutaceae potrebbero avere la priorità come produttori Pala candidato basata esclusivamente su abbondanza relativa classifica ( Tabella 2 ; [ 24 ]). Sebbene non abbiamo condotto questa analisi per i sei ASV che sono stati classificati al meglio a livello di famiglia o di ordine, alcuni di questi potrebbero valere la pena di essere considerati come potenziali produttori considerando le loro relazioni di livello superiore con i lignaggi di produzione di prodotti naturali marini. Ad esempio, gli attinobatteri marini sono associati classicamente alla produzione di numerosi prodotti naturali bioattivi (ad esempio, [ 53 , 54]), sebbene la speculazione sia difficile con gli attinobatteri SaM_ASV20 nel nucleo in quanto è solo lontanamente correlata a noti produttori di prodotti naturali. Allo stesso modo, SaM_ASV15 correlato alle Opitutaceae è classificato 7 in termini di abbondanza relativa media e rientra nella stessa famiglia il Candidatus Didemnitutus mandela associato all’ascidiana, che ospita il cluster di geni biosintetici previsto per produrre mandelalide, un polichetide glicosilato [ 55 ]. Da questo, si potrebbe dare la priorità alla Microbulbifer , Pseudovibrio e Opitutaceae ASV per le indagini a valle, con l’abbondanza relativa inferiore Nitrosomonas e NitrospiraGli ASV possiedono anche un certo potenziale, data l’abbondanza forse sorprendente del contenuto di cluster di geni biosintetici in questi taxa chemoautotrofici e generalmente di piccole dimensioni del genoma. Sebbene questa analisi si sia concentrata sulle caratterizzazioni di percorsi previsti tra generi rilevati, le distribuzioni di percorsi previsti variavano sostanzialmente tra i taxa analizzati. Il potenziale per questi nuovi ceppi associati all’ascidiana dell’Antartide di ospitare vie metaboliche secondarie rimane speculativo in quanto sono tra i componenti più variabili del genoma di un batterio
4. Conclusioni
Questo lavoro ha migliorato la nostra comprensione dell’ascidia antartica S. adareanum, Distribuzioni PalA e microbioma in diversi modi. Innanzitutto, abbiamo riscontrato che PalA è un prodotto dominante in tutti e 63 i campioni, con alcune variazioni ma nessuna tendenza coerente con l’ASV del sito, del campione o del microbioma. In secondo luogo, i risultati indicano un microbioma conservato, core, rappresentato da 21 ASV, 20 dei quali sembrano essere distinti dal batterioplancton antartico. La distribuzione filogenetica di questi taxa era diversa e distinta dagli altri microbiomi di ascide in cui sono previsti organismi con stili di vita sia eterotrofi che chemiosintetici. In terzo luogo, l’analisi della ricorrenza ha suggerito il potenziale per reti microbiche che interagiscono ecologicamente e che potrebbero migliorare la nostra comprensione di questo sistema di prodotto ascidico-microbioma-naturale. Allo stesso modo, in base al verificarsi e alla probabilità della biosintesi del prodotto naturale,ci sono un certo numero di taxa che possono avere capacità biosintetiche, incluso quello di PalA. Questi risultati fanno avanzare l’obiettivo a lungo termine della produzione di palmerolideSynoicum adareanum e ricerca sul microbioma associato all’ospite, che è costretto dal fatto che identificando il produttore, il sequenziamento del genoma potrebbe quindi fornire informazioni sulla biosintesi del PalA, e quindi portare allo sviluppo di un potenziale agente terapeutico per combattere il melanoma.
5. Materiali e metodi
5.1. Sforzo dipendente dalla coltivazione
I campioni di S. adareanum raccolti da SCUBA nel 2004 e 2007 sono stati utilizzati per la coltivazione ( Tabella S5). I campioni del 2004 sono stati archiviati in glicerolo al 20% a -80 ° C fino al trattamento mediante omogeneizzazione manuale utilizzando malta e pestello sterilizzati prima di placcare una sospensione su piastre di agar marino 2216 (BD Difco ™, Franklin Lakes, NJ, USA) nel 2006. Il I campioni del 2007 sono stati omogeneizzati immediatamente dopo la raccolta utilizzando malta e pestello sterilizzati e sono state preparate sospensioni in brodo marino 1X 2216 o acqua di mare antartica sterilizzata con filtro, quindi trasferite a 4 ° C in DRI. Poco dopo (entro 1 mese dalla raccolta), gli isolati del 2007 sono stati coltivati su tre tipi di media in cui le sospensioni inizialmente immagazzinate nel brodo marino 2216 sono state posate su agar marino (2216), mentre le preparazioni omogenate immagazzinate in acqua di mare sono state posate su media di agar VNSS [ 56] e piastre di agar di acqua di mare antartiche modificate con 3 g di estratto di lievito (BD Difco ™), 5 g di peptone (BD Difco ™) e 0,2 g di caseina idrosidata (BD Difco ™) per litro. Le colonie sono state selezionate dalle piastre iniziali e purificate attraverso tre cicli di crescita sullo stesso supporto su cui erano isolate.
5.2. Raccolte di campioni sul campo per sforzi indipendenti dalla coltivazione
Successivamente, è stata eseguita un’indagine spaziale del Synoicum adareanum in cui i campioni sono stati raccolti da SCUBA in autunno australiano tra il 23 marzo e il 3 aprile 2011. Sette siti di campionamento (profondità 24,7–31 m; Tabella S6 ) intorno alla regione accessibile dalla barca Zodiac da Palmer Sono state selezionate le stazioni in cui abbiamo campionato in un disegno nidificato in cui sono state selezionate tre colonie a lobi multipli da ciascun sito e sono stati campionati tre lobi per colonia ( Figura 1 ). In ogni sito di immersione, le colonie a lobi multipli sono state raccolte a mano in sacchi di raccolta mesh separati. Video subacquei [ 57 ] sono stati ripresi in ciascun sito, quindi sono stati osservati filmati per notare le caratteristiche generali dell’ecosistema (% di copertura delle principali specie e alghe bentoniche). In totale, 63 S. adareanumi lobi sono stati campionati (9 da ciascun sito). I campioni sono stati trasportati alla stazione di Palmer su ghiaccio e i campioni di colonie a più lobi sono stati collocati in sacchi sterili Whirl-Pak® (Nasco, Fort Atkinson, WI, USA) e congelati a -80 ° C, fino alla lavorazione a DRI e USF. Al DRI, i lobi di S. adareanum congelati peduncolati sono stati separati, quindi tagliati longitudinalmente a metà per l’elaborazione parallela attraverso condotte di rilevazione di DNA e palmerolide.
Quindi, per stabilire se la composizione del SaM fosse distinta dal batterioplancton a vita libera (frazione <2,5 μm), abbiamo considerato la sovrapposizione tra appartenenza del SaM e batterioplancton nella colonna d’acqua. A tale scopo, abbiamo utilizzato un set di dati di riferimento sull’acqua marina rappresentato da campioni raccolti nel periodo febbraio-marzo 2008 (cinque campioni) e nel periodo agosto-settembre 2008 (nove campioni) dalla stazione LTER B vicino Anvers Island (est del sito di immersione Bonaparte Point) e in alcune altre località della regione ( Tabella S7). I campioni di acqua di mare sono stati raccolti da una pompa sommergibile e da un tubo in silicone lavato con acido a 10 m nella stazione B, e usando una rosetta dotata di flaconi Niskin da 12 L per i campioni offshore a 10 e 500 m (2 campioni per profondità). I campioni di acqua di mare febbraio-marzo sono stati processati utilizzando una filtrazione in linea con un filtro da 2,5 μm (Polygard, MilliporeSigma, Burlington MA, USA) per lo screening di organismi più grandi e il batterioplancton è stato concentrato utilizzando un sistema di filtrazione a flusso tangenziale e le cellule sono state raccolte su 25 mm e filtri Supor da 0,2 μm (MilliporeSigma). I campioni di acqua di mare agosto-settembre sono stati processati usando una filtrazione in linea con un filtro da 3,0 μm (Versapor, MilliporeSigma), e quindi il batterioplancton è stato raccolto su filtri Sterivex (MilliporeSigma) da 0,2 μm usando una pompa peristaltica multicanale (Masterflex®, Cole-Parmer, Vernon Hills , I L,STATI UNITI D’AMERICA). Tutti i filtri sono stati immersi in saccarosio: Tris: buffer EDTA [58 ] e conservato congelato a -80 ° C fino all’estrazione.
5.3. Palmerolide A Screening
Sessantatre lobi di S. adareanum congelati furono tagliati a metà. Mezzi lobi sono stati liofilizzati e quindi estratti in modo esaustivo con diclorometano per tre giorni, seguito da metanolo per tre giorni. Gli estratti sono stati combinati ed essiccati su un evaporatore rotante. Gli estratti sono stati filtrati, essiccati e ricostituiti a 1,0 mg / mL per garantire che la concentrazione iniettata fosse coerente. Il residuo è stato sottoposto ad analisi liquido-cromatografia spettrometria di massa (LC – MS) usando un H 2O: gradiente ACN con acido formico costante allo 0,05%. Gli spettri di massa ad alta risoluzione sono stati registrati su uno spettrometro a tempo di volo (ESI-ToF) a ionizzazione elettrospray 6230 di Agilent Technologies. LC-MS è stato eseguito utilizzando una colonna analitica Kinetex C-18 (50 × 2,1 mm; Phenomenex, Torrance, CA, USA). La presenza di PalA è stata verificata utilizzando MS / MS su un LC-MS QTOF a massa accurata 6540 UHD Agilent Technologies.
La collezione di coltura microbica associata ad S. adareanum è stata vagliata per la presenza di PalA. Gli isolati sono stati coltivati in volumi di 30 ml e la biomassa risultante è stata liofilizzata e vagliata mediante HPLC. Ogni campione è stato analizzato in modalità ESI-SIM per masse di 585 amu e 607 amu. L’analisi cromatografica a fase inversa consisteva in un gradiente di solvente di 0,7 mL × min −1 dall’80% H 2 O: CH 3 CN al 100% CH 3 CN con acido formico 0,05% costante usando la stessa colonna C-18 di cui sopra. L’analisi è stata condotta per oltre 18 minuti. PalA ha avuto un tempo di ritenzione di circa 14 minuti. La sequenza di ventiquattro campioni è stata seguita da uno standard PalA per confermare le sue caratteristiche analitiche.
5.4. Preparazione di cellule microbiche associate a S. Adareanum
I 1-2 mm esterni del tessuto ascidico, che potrebbero contenere microrganismi associati alla superficie, sono stati rimossi usando un bisturi sterilizzato prima di sezionare i sottocampioni di tessuto (~ 0,2 g) di S. adareanum congelato (−80 ° C) (sezioni ½ lobo) . I campioni di tessuto sono stati tagliati a dadini con un bisturi sterile prima dell’omogeneizzazione in acqua sterile sterilizzata in autoclave e filtrata in acqua di mare (1 ml) in provette da 2 ml. Ogni campione è stato omogeneizzato (MiniLys, Bertin Instruments, Montigny-le-Bretonneux, Francia) usando microsfere sterili CK28 (Precellys, Bertin Instruments) 3 volte a 5000 rpm per 20 s, i campioni sono stati posti sul ghiaccio tra ogni omogeneizzazione. Gli omogenati sono stati centrifugati a 500 × ga 4 ° C per 5 minuti per eliminare i detriti di tessuto. Il surnatante è stato rimosso in una nuova provetta per un secondo giro nelle stesse condizioni. Questo surnatante è stato travasato e la sospensione cellulare centrifugata a 12.000 × g a 4 ° C per 5 minuti per raccogliere le cellule microbiche. Le sospensioni sono state immagazzinate sul ghiaccio, quindi sono entrate in una conduttura di estrazione, in cui 12 campioni sono stati processati in parallelo sul collettore QIAvac 24 Plus (Qiagen Inc., Germantown, MD, USA).
5.5. Estrazioni di DNA
Le preparazioni di cellule microbiche associate a S. adareanum sono state estratte con estrazione Powerlyzer DNEasy (Qiagen Inc.) seguendo le istruzioni del produttore a partire dall’aggiunta della soluzione di lisi. I campioni sono stati processati in parallelo in lotti di dodici alla volta utilizzando il collettore sottovuoto QiaVac 24 Plus. Due giri di lisi (5000 rpm per 60 s ciascuno con incubazione su ghiaccio) sono stati eseguiti sul MiniLys usando le perle di vetro da 0,1 mm fornite con il kit Powerlyzer. Le concentrazioni di DNA dei preparati finali sono state stimate utilizzando il rilevamento della fluorescenza del dsDNA Assay Kit (Invitrogen) Quant-iT Picogreen su uno Spectramax Gemini (Molecular Devices, Mountain View, CA, USA).
Il DNA da campioni di batterioplancton è stato estratto in seguito [ 58 ] e il DNA da colture batteriche è stato estratto utilizzando il kit DNeasy Blood and Tissue (Qiagen Inc.) seguendo le istruzioni del produttore. Tutte le concentrazioni di DNA sono state stimate usando Picogreen.
5.6. Il sequenziamento genico dell’rRNA 16S
Il sequenziamento dei tag Illumina per il microbioma di S. adareanum (SaM) ha preso di mira la regione V3 – V4 del gene 16S rRNA utilizzando primer bersaglio per procarioti 341F (5′-CCTACGGGNBGCASCAG-3 ′ [ 59 ]) e 806R (5′-GGACTACHVGGGTWTCTAAT-3 ′ [ 60]; fonte: tecnologie integrate del DNA). Il primo round di PCR ha amplificato la regione V3 – V4 utilizzando HIFI HotStart Ready Mix (Kapa Biosystems, Wilmington, MA, USA). Il primo ciclo di PCR ha utilizzato una temperatura di denaturazione di 95 ° C per 3 minuti, 20 cicli di 95 ° C per 30 secondi, 55 ° C per 30 secondi e 72 ° C per 30 secondi, seguita da un’estensione di 72 ° C per 5 minuti prima di tenerlo a 4 ° C. Il secondo round di PCR ha aggiunto sequenze di adattatori di sequenziamento specifici per Illumina e indici unici, consentendo il multiplexing, utilizzando il Nextera XT Index Kit v2 (Illumina, Inc., San Diego, CA, USA) e HIFI HotStart Ready Mix (Kapa Biosystems). Il secondo ciclo di PCR ha utilizzato una temperatura di denaturazione di 95 ° C per 3 minuti, 8 cicli di 95 ° C per 30 secondi, 55 ° C per 30 secondi e 72 ° C per 30 secondi, seguito da un prolungamento di 72 ° C per 5 minuti prima di tenerlo a 4 ° C.Gli ampliconi sono stati ripuliti usando perline AMPure XP (Beckman Coulter, Indianapolis, IN, USA). È stato elaborato un controllo di amplificazione PCR senza modello ma non ha mostrato una banda nella regione dell’amplicone V3 – V4 ed è stato sequenziato per la conferma. Un dosaggio Qubit dsDNA HS (ThermoFisher Scientific, Waltham, MA, USA) è stato utilizzato per le stime della concentrazione del DNA. La dimensione media della biblioteca è stata determinata dal kit DNA ad alta sensibilità (Agilent) e dal kit di quantificazione delle biblioteche – il kit Illumina / Universal (KAPA Biosystems) ha quantificato le librerie preparate. Il pool di ampliconi sequenziato su letture Illumina MiSeq generate a 301 bp a coppie accoppiate è stato demultiplato usando bcl2fastq di Illumina.È stato elaborato un controllo di amplificazione PCR senza modello ma non ha mostrato una banda nella regione dell’amplicone V3 – V4 ed è stato sequenziato per la conferma. Un dosaggio Qubit dsDNA HS (ThermoFisher Scientific, Waltham, MA, USA) è stato utilizzato per le stime della concentrazione del DNA. La dimensione media della biblioteca è stata determinata dal kit DNA ad alta sensibilità (Agilent) e dal kit di quantificazione delle biblioteche – il kit Illumina / Universal (KAPA Biosystems) ha quantificato le librerie preparate. Il pool di ampliconi sequenziato su letture Illumina MiSeq generate a 301 bp a coppie accoppiate è stato demultiplato usando bcl2fastq di Illumina.È stato elaborato un controllo di amplificazione PCR senza modello ma non ha mostrato una banda nella regione dell’amplicone V3 – V4 ed è stato sequenziato per la conferma. Un dosaggio Qubit dsDNA HS (ThermoFisher Scientific, Waltham, MA, USA) è stato utilizzato per le stime della concentrazione del DNA. La dimensione media della biblioteca è stata determinata dal kit DNA ad alta sensibilità (Agilent) e dal kit di quantificazione delle biblioteche – il kit Illumina / Universal (KAPA Biosystems) ha quantificato le librerie preparate. Il pool di ampliconi sequenziato su letture Illumina MiSeq generate a 301 bp a coppie accoppiate è stato demultiplato usando bcl2fastq di Illumina.La dimensione media della biblioteca è stata determinata dal kit DNA ad alta sensibilità (Agilent) e dal kit di quantificazione delle biblioteche – il kit Illumina / Universal (KAPA Biosystems) ha quantificato le librerie preparate. Il pool di ampliconi sequenziato su letture Illumina MiSeq generate a 301 bp a coppie accoppiate è stato demultiplato usando bcl2fastq di Illumina.La dimensione media della biblioteca è stata determinata dal kit DNA ad alta sensibilità (Agilent) e dal kit di quantificazione delle biblioteche – il kit Illumina / Universal (KAPA Biosystems) ha quantificato le librerie preparate. Il pool di ampliconi sequenziato su letture Illumina MiSeq generate a 301 bp a coppie accoppiate è stato demultiplato usando bcl2fastq di Illumina.
I campioni di batterioplancton inviati al Joint Genome Institute (JGI, Walnut Creek, California, USA) per la preparazione della biblioteca e il sequenziamento MiSeq Illumina a fine paia (2 × 250 bp) della regione variabile 4 (V4) mirata ai procarioti usando primer 515F ( 5′-GTGCCAGCMGCCGCGGTAA-3 ′) e 806R (5′-GGACTACHVGGGTWTCTAAT-3 ′ [ 60 ]; fonte: tecnologie integrate del DNA). I controlli di amplificazione PCR senza modello sono stati eseguiti come sopra ed erano negativi. L’elaborazione della sequenza includeva la rimozione di contaminanti PhiX e adattatori Illumina presso JGI.
L’identità degli isolati coltivati è stata confermata dal sequenziamento del gene 16S rRNA utilizzando primer Bact27F e Bact1492R o sequenziando direttamente i prodotti PCR purificati con gel di agarosio (Qiagen Inc.) o la clonazione TA (Invitrogen, ThermoFisher Scientific) di frammenti di PCR in E. coli seguendo le istruzioni del produttore, in cui sono stati sequenziati tre cloni per ciascuna libreria e i plasmidi sono stati purificati (Qiagen Inc.) presso il Nevada Genomics Center, dove il sequenziamento di Sanger è stato condotto su un ABI3700 (Applied Biosystems, Life Technologies, Foster City, CA, USA) ). Le sequenze sono state ritagliate e la qualità controllata tramite Sequencer, v. 5.1.
5.7. Analisi bioinformatica di sequenze di tag del gene 16S rRNA
Abbiamo impiegato una pipeline QIIME2 [ 61 ] utilizzando il plug-in DADA2 [ 62] per eliminare il rumore dai dati e generare matrici di occorrenza della variante di sequenza di ampliconi (ASV) per i campioni SaM e batterioplancton. Il rigore della determinazione dell’ASV è stato utilizzato in questo caso data la maggiore capacità di scoprire la variabilità nell’area di studio geografico limitata, l’interesse a scoprire modelli di specificità dell’ospite e, in ultima analisi, a identificare i membri centrali conservati del microbioma, almeno uno dei che può essere capace della biosintesi di PalA. I set di dati di sequenza sono stati inizialmente importati nel formato di lavoro QIIME2 e la qualità di forward e reverse è stata verificata. I parametri di taglio predefiniti includevano il taglio di tutte le basi dopo il primo punteggio di qualità di 2, inoltre, le prime 10 basi sono state ritagliate e le letture più brevi di 250 basi sono state scartate.Successivamente è stato utilizzato l’algoritmo DADA2 per eliminare il rumore dalle letture (corregge gli errori di sostituzione e inserimento / eliminazione e determina le varianti della sequenza). Dopo la rimozione del rumore, le letture sono state unite. Gli ASV sono stati costruiti raggruppando le esclusive sequenze de-noised complete (l’equivalente del 100% di OTU, unità tassonomiche operative). Gli ASV sono stati ulteriormente curati nella pipeline QIIME2-DADA2 rimuovendo le chimere in ciascun campione singolarmente se possono essere esattamente ricostruiti combinando un segmento sinistro e un segmento destro da due sequenze “parent” più abbondanti. Un classificatore pre-addestrato SILVA 132 99% 16S rRNA Naive Bayes (Gli ASV sono stati ulteriormente curati nella pipeline QIIME2-DADA2 rimuovendo le chimere in ciascun campione singolarmente se possono essere esattamente ricostruiti combinando un segmento sinistro e un segmento destro da due sequenze “parent” più abbondanti. Un classificatore pre-addestrato SILVA 132 99% 16S rRNA Naive Bayes (Gli ASV sono stati ulteriormente curati nella pipeline QIIME2-DADA2 rimuovendo le chimere in ciascun campione singolarmente se possono essere esattamente ricostruiti combinando un segmento sinistro e un segmento destro da due sequenze “parent” più abbondanti. Un classificatore pre-addestrato SILVA 132 99% 16S rRNA Naive Bayes (https://data.qiime2.org/2019.1/common/silva-132-99-nb-classifier.qza ) è stato utilizzato per eseguire la classificazione tassonomica. Le composizioni del SaM e degli ASV batterioplancton sono state riassunte in proporzione a diversi livelli di tassonomia, compresi i ranghi di genere, famiglia, ordine, classe e phylum. Al fine di conservare tutti i campioni per l’analisi della diversità, abbiamo impostato la frequenza di letture più bassa per campione (n = 62 campioni a 19.003 letture; n = 63 campioni a 9987 letture) come profondità di rarefazione per normalizzare i dati per le differenze nel conteggio delle sequenze. Gli ASV assegnati a Eukarya o con taxa non assegnati (sospetti contaminanti) sono stati rimossi dalla matrice di occorrenza finale in modo tale che i conteggi delle letture della matrice finale fossero leggermente irregolari con il numero più basso di letture per campione con 9961 letture.
Gli ASV SaM sono stati integrati nel core (altamente persistente) se presenti in ≥ 80% dei campioni (Core80), dinamici se presenti nel 50% -79% dei campioni (Dynamic50) e in quelli che comprendono il microbioma naturalmente fluttuante, o frazione variabile, che è stato definito come gli ASV presenti in <50% dei campioni [ 2 , 3 ]. Abbiamo usato questi raggruppamenti conservativi del microbioma centrale a causa della bassa profondità del sequenziamento nel nostro studio [ 3 ].
Le identità di ASV tra il SaM, gli isolati batterici di S. adareanum e i set di dati del batterioplancton sono state confrontate usando CD-HIT (cd-hit-est-2d; http://cd-hit.org). Il set di dati SaM più grande che includeva 19.003 sequenze per campione è stato utilizzato per questi confronti per massimizzare la capacità di identificare le corrispondenze; si noti che questo set esclude un campione, Bon1b, che aveva la metà del numero di ASV, sebbene nel complesso questo set di dati più ampio includa quasi 200 sequenze aggiuntive nella frazione Variabile per il confronto. Gli ASV con identità al 100% e al 97% tra i confronti a coppie sono stati riassunti in termini di appartenenza alle frazioni Core, Dynamic o Variable del SaM. Allo stesso modo, il CD-HIT è stato usato per dereplicare le sequenze di isolati a un livello di identità della sequenza del 99%, e quindi il set dereplicato è stato confrontato con il set di dati iTag del batterioplancton.
Analisi filogenetica degli ASV SaM, isolati batterici di S. adareanum e sequenze clonate del gene 16S rRNA da Riesenfeld et al. [ 13 ] è stato condotto rispetto alle sequenze vicine identificate nel Ribosomal Database Project e SILVA e un gruppo esterno archaea usando MEGA v.7 [ 63 ]. Sono stati costruiti due alberi di massima verosimiglianza: il primo con gli ASV Core80 e il secondo con gli ASV Core80 e Dynamic50. In entrambi gli alberi sono state utilizzate 369 posizioni allineate. Sono stati eseguiti in totale 1000 replicati bootstrap in entrambi i casi, in cui la percentuale (≥ 50%) degli alberi in cui i taxa associati raggruppati insieme sono mostrati accanto ai rami.
5.8. Analisi statistiche
I test T sono stati eseguiti in Statistica (v. 13, Tibco Software, Palo Alto, CA, USA) per determinare la significatività ( p<0,05) della variazione da sito a sito, all’interno e tra le colonie delle concentrazioni di PalA. Matrici di similarità e analisi di clustering gerarchici sono state eseguite utilizzando PRIMER v.7 e PERMANOVA + (PRIMER-e, Auckland, Nuova Zelanda). Nella maggior parte dei casi sono state eseguite analisi sul microbioma completo e sulle tre frazioni di microbioma. I dati di occorrenza di ASV sono stati trasformati in radice quadrata per tutte le analisi. Una mappa di calore basata sull’occorrenza di ASV Core80 è stata generata con i dati trasformati ed è stato impiegato il clustering gerarchico con parametro medio di gruppo, che è stato integrato con la sicurezza SIMPROF usando 9.999 permutazioni e un livello di significatività del 5%. Le matrici di somiglianza di Bray-Curtis sono state create utilizzando occorrenze ASV senza l’uso di una variabile fittizia. Per determinare i modelli di base nella struttura della comunità all’interno ditra la colonia e la variazione del sito, la significatività è stata determinata mediante t-test usando Statistica v. 13. Per confrontare la variazione tra-(n = 9) e tra-colonia (n = 27), nove valori di somiglianza a coppie tra le colonie erano campionati casualmente al fine di confrontare uguali dimensioni del campione, verificando che l’omogeneità della varianza fosse simile tra loro. Quindi è stato condotto il ridimensionamento multidimensionale metrico di soglia (tmMDS) basato sullo schema di adattamento di Kruskal 1, incluse 500 iterazioni e uno stress minimo di 0,001. Il test del profilo di somiglianza tramite SIMPROF è stato eseguito sulla base di un’ipotesi nulla secondo cui nessun gruppo avrebbe dimostrato differenze nelle occorrenze di ASV. Questo algoritmo di clustering è stato anche utilizzato per generare livelli di confidenza sul grafico MDS, che erano impostati al 65% e al 75%. Inoltre, sono state calcolate le aree bootstrap al 95% con 43 bootstrap per gruppo,impostato per garantire un minimo di 0,99 rho. Al fine di valutare il contributo di ciascun fattore alla varianza della comunità microbica in questo disegno sperimentale annidato, è stato utilizzato Site (Colony), analisi multivariata permutazionale della varianza (PERMANOVA). Sono stati calcolati i centroidi basati sul sito e l’algoritmo PERMDISP è stato utilizzato per determinare il grado di dispersione attorno al centroide per ciascun sito. Nel complesso, è stata determinata la differenza di dispersione da sito a sito e sono stati calcolati anche confronti a coppie, con 9999 permutazioni utilizzate per determinare la significatività (P (perm) <0,05). L’analisi esplorativa dei principali contribuenti dell’ASV alla somiglianza è stata eseguita utilizzando la procedura SIMPER basata su siti e colonie, con un taglio per i contributi bassi fissato al 70%.Al fine di valutare il contributo di ciascun fattore alla varianza della comunità microbica in questo disegno sperimentale annidato, è stato utilizzato Site (Colony), analisi multivariata permutazionale della varianza (PERMANOVA). Sono stati calcolati i centroidi basati sul sito e l’algoritmo PERMDISP è stato utilizzato per determinare il grado di dispersione attorno al centroide per ciascun sito. Nel complesso, è stata determinata la differenza di dispersione da sito a sito e sono stati calcolati anche confronti a coppie, con 9999 permutazioni utilizzate per determinare la significatività (P (perm) <0,05). L’analisi esplorativa dei principali contribuenti dell’ASV alla somiglianza è stata eseguita utilizzando la procedura SIMPER basata su siti e colonie, con una riduzione per contributi bassi fissata al 70%.Al fine di valutare il contributo di ciascun fattore alla varianza della comunità microbica in questo disegno sperimentale annidato, è stato utilizzato Site (Colony), analisi multivariata permutazionale della varianza (PERMANOVA). Sono stati calcolati i centroidi basati sul sito e l’algoritmo PERMDISP è stato utilizzato per determinare il grado di dispersione attorno al centroide per ciascun sito. Nel complesso, è stata determinata la differenza di dispersione da sito a sito e sono stati calcolati anche confronti a coppie, con 9999 permutazioni utilizzate per determinare la significatività (P (perm) <0,05). L’analisi esplorativa dei principali contribuenti dell’ASV alla somiglianza è stata eseguita utilizzando la procedura SIMPER basata su siti e colonie, con una riduzione per contributi bassi fissata al 70%.è stata utilizzata l’analisi multivariata permutazionale della varianza (PERMANOVA). Sono stati calcolati i centroidi basati sul sito e l’algoritmo PERMDISP è stato utilizzato per determinare il grado di dispersione attorno al centroide per ciascun sito. Nel complesso, è stata determinata la differenza di dispersione da sito a sito e sono stati calcolati anche confronti a coppie, con 9999 permutazioni utilizzate per determinare la significatività (P (perm) <0,05). L’analisi esplorativa dei principali contribuenti dell’ASV alla somiglianza è stata eseguita utilizzando la procedura SIMPER basata su siti e colonie, con una riduzione per contributi bassi fissata al 70%.è stata utilizzata l’analisi multivariata permutazionale della varianza (PERMANOVA). Sono stati calcolati i centroidi basati sul sito e l’algoritmo PERMDISP è stato utilizzato per determinare il grado di dispersione attorno al centroide per ciascun sito. Nel complesso, è stata determinata la differenza di dispersione da sito a sito e sono stati calcolati anche confronti a coppie, con 9999 permutazioni utilizzate per determinare la significatività (P (perm) <0,05). L’analisi esplorativa dei principali contribuenti dell’ASV alla somiglianza è stata eseguita utilizzando la procedura SIMPER basata su siti e colonie, con una riduzione per contributi bassi fissata al 70%.con 9999 permutazioni utilizzate per determinare la significatività (P (perm) <0,05). L’analisi esplorativa dei principali contribuenti dell’ASV alla somiglianza è stata eseguita utilizzando la procedura SIMPER basata su siti e colonie, con una riduzione per contributi bassi fissata al 70%.con 9999 permutazioni utilizzate per determinare la significatività (P (perm) <0,05). L’analisi esplorativa dei principali contribuenti dell’ASV alla somiglianza è stata eseguita utilizzando la procedura SIMPER basata su siti e colonie, con una riduzione per contributi bassi fissata al 70%.
Le reti di ricorrenza sono state costruite utilizzando set di dati di occorrenza ASV filtrati in cui gli ASV sono stati filtrati solo a quelli che erano presenti in almeno cinque campioni risultanti in un set di dati ASV 102. La matrice 102 × 63 è stata fornita come input per FlashWeave v1.0 [ 64 ] utilizzando i parametri predefiniti e visualizzata in Gephi v. 0.9.2 [ 65 ]. Quindi, per considerare se gli ASV nelle frazioni Core80, Dynamic50 o Variable del SaM fossero affiliati a particolari livelli di PalA nei lobi di ascidiana, l’ottimale robusto e la gamma di nicchia PalA sono stati calcolati utilizzando i dati contestuali normalizzati in base al peso a secco [ 66 ] ]. Analisi della rete di correlazione genica ponderata (pacchetto WGCNA in R [ 67]) è stato utilizzato per identificare i moduli e la loro correlazione con i livelli di PalA. La matrice è stata normalizzata a somma totale [ 68 ] e WGCNA è stato utilizzato in modalità firmata. Sono stati rilevati pochi moduli, sebbene non fossero correlati con PalA. I moduli sono stati proiettati sulla rete di ricorrenza FlashWeave e chiamati sottosistemi.
5.9. Analisi del cluster di geni biosintetici
Successivamente, al fine di prevedere la probabilità dei lignaggi ASV Core80 che ospitano il potenziale per la biosintesi di prodotti naturali, abbiamo progettato una meta-analisi dei genomi vicini trovati nel database dei genomi microbici integrati (IMG) [ 69 ]. L’analisi è stata condotta solo per gli ASV in cui la fiducia nell’assegnazione tassonomica era a livello di genere. Pertanto, sono stati raccolti genomi da IMG associati a un totale di 9 generi ( Microbulbifer (16 genomi), Pseudovibrio (24 genomi), Endozoicomonas (11 genomi), Nitrosomonas (19 su 68 genomi totali in questo genere), Nitrospira (14 genomi), Hoeflea (7 genomi), Lutibacter(12 genomi), Halocynthilibacter (2 genomi) e nel caso dei Lentimonas , poiché non sono stati trovati genomi, abbiamo raccolto 8 genomi dalla famiglia delle Puniciococcaceae ). Ciò si traduce in 113 genomi che sono stati sottoposti ad analisi antiSMASH [ 70 ]. Sono stati tabulati i genomi e la conta dei gruppi di geni biosintetici assegnati alla peptide sintasi nonribosomiale, alla polichetide sintasi o ad un ibrido delle due classi ( Tabella S4 ).
5.10. Disponibilità dei dati
Il microbioma di Synoicum adareanum Le informazioni sulla sequenza Illumina e i metadati associati sono descritti nel NCBI BioProject PRJNA597083 ( https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA597083 ) e le sequenze del gene 16S rRNA della collezione di colture di S. adareanum erano depositato in GenBank sotto MN960541- MN960556. Le informazioni sulla sequenza batterioplancton e i metadati associati sono descritti in NCBI BioProject PRJNA602715 ( https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA602715 ). Metadati del progetto antartico [ 71 ] e metadati ambientali (identificatori del set di dati: Synoicum_adareanum _microbiome_part1, Synoicum_adareanum _microbiome_part2, Synoicum_adareanum_microbiome_part3) sono disponibili su: POLA 3 R (2020) —POLA 3 R Polar Links to Antarctic, Arctic and Alpine Research. Comitato scientifico per la ricerca antartica, portale antartico sulla biodiversità, ( www.biodiversity.aq/pola3r/ ).
Materiali supplementari
Di seguito sono disponibili online all’indirizzo https://www.mdpi.com/1660-3397/18/6/298/s1 , Tabella S1: occorrenze, sequenze e affiliazioni tassonomiche di ASV, Tabella S2: stimatori PERMANOVA dei driver di variabilità, Tabella S3: distribuzione tassonomica degli ASV di batterioplancton, tabella S4: gruppi di geni biosintetici nei genomi batterici correlati ai generi Core80 SaM, tabella S5: collezioni di Synoicum adareanum e preparati per la coltivazione di microbiomi, tabella S6: Synoicum adareanumraccolte per la caratterizzazione del palmerolide A e del microbioma mediante sequenziamento del tag del gene rRNA V3 – V4, Tabella S7: collezioni di batteriotoplancton utilizzate nel sequenziamento del tag del gene r4 dell’RRNA. Figura S1: Risultati dei test t a coppie dei livelli di PalA determinati dalla spettrometria di massa, Figura S2: Albero filogenetico del gene rRNA 16S con massima probabilità, Figura S3: Somiglianza media a coppie media all’interno e tra le strutture comunitarie del microbioma di S. adareanum , Figura S4: grafici tmMDS che rappresentano il microbioma dei 63 campioni di S. adareanum e la Figura S5. Nicchia PalA ottimale per ASV di microbiomi di S. adareanum.
Contributi dell’autore
Questo lavoro è stato il risultato di uno sforzo di gruppo in cui sono stati riconosciuti i seguenti contributi: concettualizzazione, metodologia e sperimentazione AEM, PSGC e BJB, AEM, NEA, LB, DE, MLH, SK, CSR e RMY; validazione, analisi formale KWD, MLH e BJB, AEM, NEA, ED, DE, SK e C.-CL; cura dei dati, MLH e C.-CL; stesura — preparazione di bozze originali, AEM, BJB, NEA, DE, SK e C.-CL; scrittura: revisione e modifica, AEM, NEA, BJB, LB, PSGC, AEKD, DE, SK, CSR e RMY; supervisione, AEM, BJB, PSGC, KWD, AEKD e CSR; amministrazione del progetto, KWD; acquisizione di finanziamenti, AEM, PSGC e BJB Tutti gli autori hanno letto e accettato la versione pubblicata del manoscritto.
finanziamento
Il supporto per questa ricerca è stato fornito in parte dal premio National Institute of Health (CA205932) a AEM, BJB e PSGC, con il supporto aggiuntivo dei premi della National Science Foundation (OPP-0442857, ANT-0838776 e PLR-1341339 a BJB, ANT-0632389 al premio DEM-0532893 alla CSR dell’AEM e della borsa di ricerca post-dottorato. Il supporto per il sequenziamento del batterioplancton è stato fornito nell’ambito del Programma di sequenziamento comunitario del Joint Genome Institute (da JGI-634 a AEM).
Ringraziamenti
È riconosciuta l’assistenza di numerosi collaboratori e studenti tra cui Charles Amsler, Margaret Amsler, Jason Cuce, Bill Dent, Alex Dussaq, Cheryl Gleasner, Alan Maschek, Robert W. Read, Andrew Shilling, Santana Thomas e lo staff di supporto scientifico della Stazione Palmer .
Conflitto di interessi
Gli autori dichiarano assenza di conflitto di interesse.
Riferimenti
Taylor, MW; Tsai, P .; Simister, RL; Deines, P .; Botte, E .; Ericson, G .; Schmitt, S .; Webster, i batteri NS “specifici per la spugna” sono diffusi (ma rari) in diversi ambienti marini. ISME J. 2013 , 7 , 438–443. [ Google Scholar ] [ CrossRef ] [ PubMed ]
Ainsworth, TD; Krause, L .; Bridge, T .; Torda, G .; Raina, JB; Zakrzewski, M .; Gates, RD; Padilla-Gamino, JL; Spalding, HL; Smith, C .; et al. Il microbioma del nucleo di corallo identifica i taxa batterici rari come onosimbi ubiquitari. ISME J. 2015 , 9 , 2261–2274. [ Google Scholar ] [ CrossRef ] [ PubMed ]
Hernandez-Agreda, A .; Leggat, W .; Bongaerts, P .; Ainsworth, TD La firma microbica fornisce informazioni sulle basi meccanicistiche del successo dei coralli negli habitat di barriera. mBio 2016 , 7 , e00560-16. [ Google Scholar ] [ CrossRef ] [ PubMed ]
Burgsdorf, I .; Erwin, PM; Lopez-Legentil, S .; Cerrano, C .; Haber, M .; Frenk, S .; Steindler, L. Biogeografia piuttosto che associazione con strutture cianobatteriche comunità microbiche simbiotiche nella spugna marina Petrosia ficiformis . Davanti. Microbiol. 2014 , 5 , 529. [ Google Scholar ] [ CrossRef ] [ PubMed ]
Kelly, LW; Williams, GJ; Barott, KL; Carlson, CA; Dinsdale, EA; Edwards, RA; Haas, AF; Haynes, M .; Lim, YW; McDole, T .; et al. Adattamento genomico locale dei microbiomi associati alla barriera corallina a gradienti di variabilità naturale e fattori di stress antropogenici. Proc. Natl. Acad. Sci. Stati Uniti d’America 2014 , 111 , 10227-10232. [ Google Scholar ] [ CrossRef ]
Pantos, O .; Bongaerts, P .; Dennis, PG; Tyson, GW; Hoegh-Guldberg, O. Le condizioni ambientali specifiche dell’habitat controllano principalmente i microbiomi del corallo Seriatopora hystrix . ISME J. 2015 , 9 , 1916-1927. [ Google Scholar ] [ CrossRef ]
Lo Guidice, A .; Azzaro, M .; Schiaparelli, S. Symbionts microbici di invertebrati bentonici marini antartici. Nel ruolo ecologico dei microrganismi nell’ambiente antartico ; Castro-Sowinski, S., Ed .; Springer Polar Sciences, Springer Nature: Basilea, Svizzera, 2019; pagg. 277–296. [ Google Scholar ]
Cardenas, CA; Gonzalez-Aravena, M .; Font, A .; Hestetun, JT; Hajdu, E .; Trefault, N .; Malmbergg, M .; Bongcarn-Rudloff, E. Elevata somiglianza nel microbiota delle spugne di acqua fredda del genere Mycale da due diverse aree geografiche. PeerJ 2018 , 6 . [ Google Scholar ] [ CrossRef ]
Webster, NS; Negri, AP; Munro, M .; Battershill, CN Diverse comunità microbiche abitano spugne antartiche. Environ. Microbiol. 2004 , 6 , 288–300. [ Google Scholar ] [ CrossRef ]
Steinert, G .; Wemheuer, B .; Janussen, D .; Erpenbeck, D .; Daniel, R .; Simon, M .; Brinkhoff, T .; Schupp, PJ Diversità procariotica e modelli di comunità nelle spugne continentali antartiche. Davanti. Mar. Sci. 2019 , 6 , 297. [ Google Scholar ] [ CrossRef ]
Webster, NS; Bourne, D. Struttura della comunità batterica associata al corallo molle antartico, Alcyonium antarcticum . FEMS Microbiol. Ecol. 2007 , 59 , 81–94. [ Google Scholar ] [ CrossRef ]
Murray, AE; Rack, FR; Zook, R .; Williams, MJM; Higham, ML; Broe, M .; Kaufmann, RS; Daly, M. Composizione del microbioma e diversità dell’anemone di mare, Edwardsiella andrillae . Integr. Comp. Biol. 2016 , 56 , 542–555. [ Google Scholar ] [ CrossRef ] [ PubMed ]
Riesenfeld, C.S.; Murray, A.E.; Baker, B.J. Characterization of the microbial community and polyketide biosynthetic potential in the Palmerolide-producing tunicate, Synoicum adareanum. J. Nat. Prod.2008, 71, 1812–1818. [Google Scholar] [CrossRef] [PubMed]
Palermo, J.A.; Brasco, M.F.R.; Spagnuolo, C.; Seldes, A.M. Illudalane sesquiterpenoids from the soft coral Alcyonium paessleri: The first natural nitrate esters. J. Org. Chem.2000, 65, 4482–4486. [Google Scholar] [CrossRef] [PubMed]
Diyabalanage, T.; Amsler, C.D.; McClintock, J.B.; Baker, B.J. Palmerolide A, a cytotoxic macrolide from the Antarctic tunicate Synoicum adareanum. J. Am. Chem. Soc.2006, 128, 5630–5631. [Google Scholar] [CrossRef] [PubMed]
Erwin, P.M.; Pineda, M.C.; Webster, N.; Turon, X.; Lopez-Legentil, S. Down under the tunic: Bacterial biodiversity hotspots and widespread ammonia-oxidizing archaea in coral reef ascidians. ISME J.2014, 8, 575–588. [Google Scholar] [CrossRef] [PubMed]
Lopez-Legentil, S.; Turon, X.; Espluga, R.; Erwin, P.M. Temporal stability of bacterial symbionts in a temperate ascidian. Front. Microbiol.2015, 6, 1022. [Google Scholar] [CrossRef]
Evans, J.S.; Erwin, P.M.; Shenkar, N.; Lopez-Legentil, S. Introduced ascidians harbor highly diverse and host-specific symbiotic microbial assemblages. Sci. Rep.2017, 7, 11033. [Google Scholar] [CrossRef]
Evans, J.S.; Erwin, P.M.; Shenkar, N.; Lopez-Legentil, S. A comparison of prokaryotic symbiont communities in nonnative and native ascidians from reef and harbor habitats. FEMS Microbiol. Ecol.2018, 94, fiy139. [Google Scholar] [CrossRef]
Cahill, P.L.; Fidler, A.E.; Hopkins, G.A.; Wood, S.A. Geographically conserved microbiomes of four temperate water tunicates. Environ. Microbiol. Rep.2016, 8, 470–478. [Google Scholar] [CrossRef]
Dror, H.; Novak, L.; Evans, J.S.; Lopez-Legentil, S.; Shenkar, N. Core and dynamic microbial communities of two invasive ascidians: Can host-symbiont dynamics plasticity affect invasion capacity? Microb. Ecol.2019, 78, 170–184. [Google Scholar] [CrossRef]
Erwin, P.M.; Pineda, M.C.; Webster, N.; Turon, X.; Lopez-Legentil, S. Small core communities and high variability in bacteria associated with the introduced ascidian Styela plicata. Symbiosis2013, 59, 35–46. [Google Scholar] [CrossRef]
Blasiak, L.C.; Zinder, S.H.; Buckley, D.H.; Hill, R.T. Bacterial diversity associated with the tunic of the model chordate Ciona intestinalis. ISME J.2014, 8, 309–320. [Google Scholar] [CrossRef] [PubMed]
Tianero, M.D.B.; Kwan, J.C.; Wyche, T.P.; Presson, A.P.; Koch, M.; Barrows, L.R.; Bugni, T.S.; Schmidt, E.W. Species specificity of symbiosis and secondary metabolism in ascidians. ISME J.2015, 9, 615–628. [Google Scholar] [CrossRef] [PubMed]
Peng, X.; Adachi, K.; Chen, C.Y.; Kasai, H.; Kanoh, K.; Shizuri, Y.; Misawa, N. Discovery of a marine bacterium producing 4-hydroxybenzoate and its alkyl esters, parabens. Appl. Environ. Microbiol.2006, 72, 5556–5561. [Google Scholar] [CrossRef]
Fukunaga, Y.; Kurahashi, M.; Tanaka, K.; Yanagi, K.; Yokota, A.; Harayama, S. Pseudovibrio ascidiaceicola sp. nov., isolated from ascidians (sea squirts). Int. J. Syst. Evol. Microbiol.2006, 56, 343–347. [Google Scholar] [CrossRef]
Schreiber, L.; Kjeldsen, K.U.; Funch, P.; Jensen, J.; Obst, M.; Lopez-Legentil, S.; Schramm, A. Endozoicomonas are specific, facultative symbionts of sea squirts. Front. Microbiol.2016, 7, 1042. [Google Scholar] [CrossRef]
Gonzalez, J.M.; Mayer, F.; Moran, M.A.; Hodson, R.E.; Whitman, W.B. Microbulbifer hydrolyticus gen.nov., sp. nov., and Marinobacterium georgiense gen.nov.sp.nov., two marine bacteria from a lignin-rich pulp mill waste enrichment community. Int. J. Syst. Evol. Microbiol.1997, 47, 369–376. [Google Scholar]
Schmidt, E.W.; Donia, M.S. Life in cellulose houses: Symbiotic bacterial biosynthesis of ascidian drugs and drug leads. Curr. Opin. Biotechnol.2010, 21, 827–833. [Google Scholar] [CrossRef]
Dou, X.; Dong, B. Origins and bioactivities of natural compounds derived from marine ascidians and their symbionts. Mar. Drugs2019, 17, 670. [Google Scholar] [CrossRef]
Little, A.E.; Robinson, C.J.; Peterson, S.B.; Raffa, K.F.; Handelsman, J. Rules of engagement: Interspecies interactions that regulate microbial communities. Annu. Rev. Microbiol.2008, 62, 375–401. [Google Scholar] [CrossRef]
Imamura, N.; Nishuma, M.; Takader, T.; Adachi, K.; Sakai, M.; Sano, H. New anticancer antibiotics pelagiomicins, produced by a new marine bacterium Pelagiobacter variabilis. J. Antibiot.1997, 50, 8–12. [Google Scholar] [CrossRef]
Quevrain, E.; Domart-Coulon, I.; Pernice, M.; Bourguet-Kondracki, M.L. Novel natural parabens produced by a Microbulbifer bacterium in its calcareous sponge host Leuconia nivea. Environ. Microbiol.2009, 11, 1527–1539. [Google Scholar] [CrossRef]
Martínez-García, M.; Stief, P.; Díaz-Valdés, M.; Wanner, G.; Ramos-Esplá, A.; Dubilier, N.; Antón, J. Ammonia-oxidizing Crenarchaeota and nitrification inside the tissue of a colonial ascidian. Environ. Microbiol.2008, 10, 2991–3001. [Google Scholar]
Hatzenpichler, R. Diversity, physiology, and niche differentiation of ammonia-oxidizing archaea. Appl. Environ. Microbiol.2012, 78, 7501–7510. [Google Scholar] [CrossRef]
Murray, A.E.; Preston, C.M.; Massana, R.; Taylor, L.T.; Blakis, A.; Wu, K.; DeLong, E.F. Seasonal and spatial variability of bacterial and archaeal assemblages in the coastal waters off Anvers Island, Antarctica. Appl. Environ. Microbiol.1998, 64, 2585–2595. [Google Scholar] [CrossRef]
Murray, A.E.; Grzymski, J.J. Diversity and genomics of Antarctic marine micro-organisms. Phil. Trans. Roy. Soc. B Biol. Sci.2007, 362, 2259–2271. [Google Scholar] [CrossRef]
Grzymski, J.J.; Riesenfeld, C.S.; Williams, T.J.; Dussaq, A.M.; Ducklow, H.; Erickson, M.; Cavicchioli, R.; Murray, A.E. A metagenomic assessment of winter and summer bacterioplankton from Antarctica Peninsula coastal surface waters. ISME J.2012, 6, 1901–1915. [Google Scholar] [CrossRef]
Rhoades, D. Evolution of Plant Chemical Defense Against Herbivores. Herbivores: Their Interaction with Secondary Plant Metabolites; Academic Press: New York, NY, USA, 1979. [Google Scholar]
McPhail, K.L.; Davies-Coleman, M.T.; Starmer, J. Sequestered chemistry of the Arminacean nudibranch Leminda millecra in Algoa Bay, South Africa. J. Nat. Prod.2001, 64, 1183–1190. [Google Scholar] [CrossRef]
Carbone, M.; Gavagnin, M.; Haber, M.; Guo, Y.W.; Fontana, A.; Manzo, E.; Genta-Jouve, G.; Tsoukatou, M.; Rudman, W.B.; Cimino, G.; et al. Packaging and delivery of chemical weapons: A defensive trojan horse stratagem in chromodorid nudibranchs. PLoS ONE2013, 8, e62075. [Google Scholar] [CrossRef]
Schupp, P .; Eder, C .; Paul, V .; Proksch, P. Distribuzione dei metaboliti secondari nella spugna Oceanapia sp. e le sue implicazioni ecologiche. Mar. Biol. 1999 , 135 , 573-580. [ Google Scholar ] [ CrossRef ]
Freeman, CJ; Gleason, DF Difese chimiche, qualità nutrizionale e componenti strutturali in tre specie di spugne: Ircinia felix , I. campana e Aplysina fulva . Mar. Biol. 2010 , 157 , 1083-1093. [ Google Scholar ] [ CrossRef ]
Roue, M.; Domart-Coulon, I.; Ereskovsky, A.; Dejediat, C.; Pererz, T.; Bourguet-Kondracki, M.-L. Cellular localization of clathridimine, an antimicrobial 2-aminoimidazole alkaloid produced by the Mediterranean calcareous sponge Clathrina clathrus. J. Nat. Prod.2010, 73, 1277–1282. [Google Scholar] [CrossRef]
Furrow, F.B.; Amsler, C.D.; McClintock, J.B.; Baker, B.J. Surface sequestration of chemical feeding deterrents in the Antarctic sponge Latrunculia apicalis as an optimal defense against sea star spongivory. Mar. Biol.2003, 143, 443–449. [Google Scholar] [CrossRef]
Becerro, M.A.; Paul, V.J.; Starmer, J. Intracolonial variation in chemical defenses of the sponge Cacospongia sp. and its consequences on generalist fish predators and the specialist nudibranch predator Glossodoris pallida. Mar. Ecol. Prog. Ser.1998, 168, 187–196. [Google Scholar] [CrossRef]
Siriak, T.; Intaraksa, N.; Kaewsuwan, S.; Yuenyongsawad, S.; Suwanborirux, K.; Plubrukarn, A. Intracolonial allocation of tisoxazole macrolides in the sponge Pachastrissa nux. Chem. Biodivers.2011, 8, 238–246. [Google Scholar]
Richardson, A.D.; Aalbersberg, W.; Ireland, C.M. The patellazoles inhibit protein synthesis at nanomolar concentrations in human colon tumor cells. Anti Cancer Drugs2008, 16, 533–541. [Google Scholar] [CrossRef]
Kwan, J.C.; Donia, M.S.; Han, A.W.; Hirose, E.; Haygood, M.G.; Schmidt, E.W. Genome streamlining and chemical defense in a coral reef symbiosis. Proc. Natl. Acad. Sci. USA2012, 109, 20655–20660. [Google Scholar] [CrossRef]
Gouiffes, D.; Juge, M.; Grimaud, N.; Welin, L.; Sauviat, M.; Barbin, Y.; Laurent, D.; Roussakis, C.; Henichart, J.; Verbist, J. Bistramide A, a new toxin from the urochordata Lissoclinum bistratum Sluiter: Isolation and preliminary characterization. Toxicon1988, 26, 1129–1136. [Google Scholar] [CrossRef]
Schmidt, E.W. The secret to a successful relationship: Lasting chemistry between ascidians and their symbiotic bacteria. Invertebr. Biol.2015, 134, 88–102. [Google Scholar] [CrossRef]
Ziemert, N.; Lechner, A.; Wietz, M.; Millan-Aguinaga, N.; Chavarria, K.L.; Jensen, P.R. Diversity and evolution of secondary metabolism in the marine actinomycete genus Salinispora. Proc. Natl. Acad. Sci. USA2014, 111, E1130–E1139. [Google Scholar] [CrossRef]
Holmstrom, C.; James, S.; Neilan, B.A.; White, D.C.; Kjelleberg, S. Pseudoalteromonas tunicata sp. nov., a bacterium that produces antifouling agents. Int. J. Syst. Bacteriol.1998, 48, 1205–1212. [Google Scholar] [CrossRef]
Baker, B.J.; Dent, B. Synoicum adareanum sampling underwater video March 2011 Palmer Station Antarctica, V3. Dryad Dataset2020. Available online: https://doi.org/10.5061/dryad.gxd2547gw (accessed on 30 May 2020).
Massana, R.; Murray, A.E.; Preston, C.M.; DeLong, E.F. Vertical distribution and phylogenetic characterization of marine planktonic Archaea in the Santa Barbara Channel. Appl. Environ. Microbiol.1997, 63, 50–56. [Google Scholar] [CrossRef]
Takahashi, S.; Tomita, J.; Nishioka, K.; Hisada, T.; Nishijima, M. Development of a prokaryotic universal primer for simultaneous analysis of Bacteria and Archaea using next-generation sequencing. PLoS ONE2014, 9, e105592. [Google Scholar] [CrossRef]
Caporaso, J.G.; Lauber, C.; Walters, W.; Berg-Lyons, D.; Lozupone, C.; Turnbaugh, P.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA2011, 108, 4516–4522. [Google Scholar] [CrossRef]
Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol.2019, 37, 852–857. [Google Scholar] [CrossRef]
Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger data sets. Mol. Biol. Evol.2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
Tackmann, J.; Matias Rodrigues, J.F.; von Mering, C. Rapid inference of direct interactions in large-scale ecological networks from heterogeneous microbial sequencing data. Cell Syst.2019, 9, 286–296.e8. [Google Scholar] [CrossRef] [PubMed]
Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An Open Source Software for Exploring and Manipulating Networks. In Proceedings of the International AAAI ICWSM Conference on weblogs and social media, San Jose, CA, USA, 17–20 May 2009. [Google Scholar]
Cristóbal, E.; Ayuso, S.V.; Justel, A.; Toro, M. Robust optima and tolerance ranges of biological indicators: A new method to identify sentinels of global warming. Ecol. Res.2013, 29, 55–68. [Google Scholar] [CrossRef]
Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform.2008, 9, 559. [Google Scholar] [CrossRef]
Blin, K.; Medema, M.H.; Kottmann, R.; Lee, S.Y.; Weber, T. The antiSMASH database, a comprehensive database of microbial secondary metabolite biosynthetic gene clusters. Nucleic Acids Res.2017, 45, D555–D559. [Google Scholar] [CrossRef]
Murray, A.; Baker, B. Synoicum adareanum Microbiome. SCAR—Microbial Antarctic Resource System Metadata Dataset. , 2020. Available online: https://doi.org/10.15468/aewoib (accessed on 30 May 20
Figura 1. Mappa batimetrica dell’area di studio al largo dell’isola di Anvers. I siti di raccolta di Synoicum adareanum sono mostrati con triangoli rossi. La mappa è stata generata da Environmental Research and Assessment, Cambridge, Regno Unito, utilizzando i dati batimetrici di Arthur Harbor dal progetto di sondaggio PRIMO 2004-2006 (Dr. Scott Gallagher e Dr. Vernon Asper). Inserto: ascidiana coloniale, S. adareanum , che si presenta in gruppi di lobi multipli collegati da un peduncolo che insieme formano una colonia sul fondo del mare, raccolti a profondità che vanno dai 24 ai 31 m.
Figura 2. Rilevazione di Palmerolide A (PalA) in holobionts di S. adareanum. ( a ) Cromatogramma ionico totale derivato dal campione Lit-1a. Il picco di PalA domina la frazione diclorometano-metanolo dell’estratto di S. adareanum . Inserto: struttura PalA. ( b ) Spettro di massa di PalA (campione Lit-1a) derivato dal picco al numero di scansione 631 che mostra [M – H 2 O + H] + (C 33 H 47 N 2 O 6 calcolato m / z 567.3429), [M + H] + (C 33 H 48 N 2 O 7calcolato m / z 585.3534) e [M + Na] + (C 33 H 47 N 2 O 7 Na calcolato m / z 607.3359). ( c ) Livelli di PalA normalizzati al peso a secco dei tessuti rilevato dalla spettrometria di massa in S. adareanumtessuti holobiont (tre lobi per colonia) esaminati in tre colonie per sito attraverso l’arcipelago dell’isola di Anvers. L’errore rappresenta la replica tecnica del singolo lobo (deviazione standard). Le colonie con differenze significative nei livelli di PalA all’interno di un sito (ad esempio, Jan-1: Jan-2) sono indicate con triangoli, in cui la direzione del punto indica una colonia significativamente più alta o più bassa. I triangoli riempiti indicano significato ( p <0,05) rispetto alle altre due colonie, mentre i triangoli aperti sono diversi da una sola delle due colonie. La maggior parte delle colonie presentava differenze significative da lobo a lobo nella concentrazione di PalA e sono state osservate alcune differenze a livello di sito ( Figura S1 ).
Figura 3. Mappa del calore dei dati di occorrenza della variante di sequenza di ampliconi trasformati con radice quadrata (ASV) per il microbioma centrale. Gli ASV (ordinati e numerati) sono mostrati sull’asse y e i campioni gerarchicamente raggruppati (63) sono mostrati sull’asse x (basato sul sito; dati di abbondanza trasformati con radice quadrata). I nodi con cluster significativi sono indicati da sinistra a destra ( p <0,05); l’ordine del clustering all’interno del nodo non era significativo. La linea orizzontale tracciata sotto SaM_ASV6 delimita gli ASV presenti in tutti i 63 campioni.
Figura 4. Rapporti di somiglianza tra i campioni di microbioma di S. adareanum nell’arcipelago dell’isola di Anvers. ( a ) tmMDS di somiglianze Bray-Curtis di radice quadrata trasformato dati di occorrenza di ASV che rappresentano il microbioma dei campioni del lobo di 63 S. adareanum usando i profili di occorrenza completi di ASV. Vengono mostrati campioni di microbiomi con livelli significativi di somiglianza (vedi legenda). ( b ) la β-diversità tra i siti dell’Arcipelago di Anvers Island rappresentata da PERMDISP (9999 permutazioni) rivela differenze tra il nucleo altamente persistente, le porzioni dinamiche e variabili del S. adareanummicrobioma (errore standard mostrato). Il grado di dispersione (varianza) attorno al centroide cambia in modo significativo ( p <0,0001) per le diverse classificazioni del microbioma. I livelli più bassi di dispersione si trovano nel microbioma centrale.
Figura 5. Rete di ricorrenza ASV. Il più grande componente connesso della rete di ricorrenza (seminato con ASV trovati in almeno 5 campioni, 102 in totale) ha identificato tre sottosistemi. I colori dei nodi rappresentano le frazioni del microbioma (Core80, verde; Dynamic50, blu; Variabile, rosa). Le identità tassonomiche degli ASV sono mostrate accanto ai nodi, con il taxon di livello più alto identificato identificato.
Tabella 1. Proporzioni relative (deviazioni standard +/- +/−, n = 63) di phyla (e classe per i proteobatteri) tra le diverse frazioni di microbiomi.
Tabella 2. Affiliazioni tassonomiche del microbioma centrale, rango di abbondanza relativa e potenziale del genere affiliato nella biosintesi del cluster di geni del prodotto naturale. La tassonomia è indicata secondo la classificazione del database di tassonomia del genoma (GTDB) e la tassonomia dell’NCBI è inclusa (GTDB / NCBI) dove differiscono. Il potenziale biosintetico calcolato solo per gli ASV con assegnazioni tassonomiche a livello di genere era basato sul contenuto rappresentativo di cluster di geni biosintetici del genoma nello stesso genere (vedere la Figura S3 per l’elenco dei genomi). ASV in grassetto classificato tra i primi 10. Laddove è stato trovato più di un ASV per genere, sono state sommate l’abbondanza relativa media e le deviazioni standard. n = 63 individui.