

Coronavirus, ceppi, mutazioni.
Esistono sul pianeta 7 tipi noti di Coronavirus che possono infettare gli individui umani (Human Coronavirus), causando patologie di varia gravità media tra i soggetti colpiti. A scanso di equivoci, vale la pena indicarli:
- Sars-CoV-2 che provoca Covid-19, la patologia che attualmente risulta letale per un paio di punti percentuali sulla popolazione mondiale
- Sars-CoV che provoca la Sars, la sindrome respiratoria acuta grave, che ha un tasso di letalità del 10%, ma la cui catena di trasmissione è stata interrotta
- Mers-CoV che provoca la Mers, la sindrome respiratoria mediorientale, trasmessa per lo più dai cammelli con un tasso di letalità del 34%
- HCoVNL–63 che provoca una sindrome respiratoria lieve
- HCoV-229E che provoca una sindrome respiratoria lieve
- HCoV-OC43 che provoca una sindrome respiratoria lieve
- HKU1 che provoca una sindrome respiratoria lieve
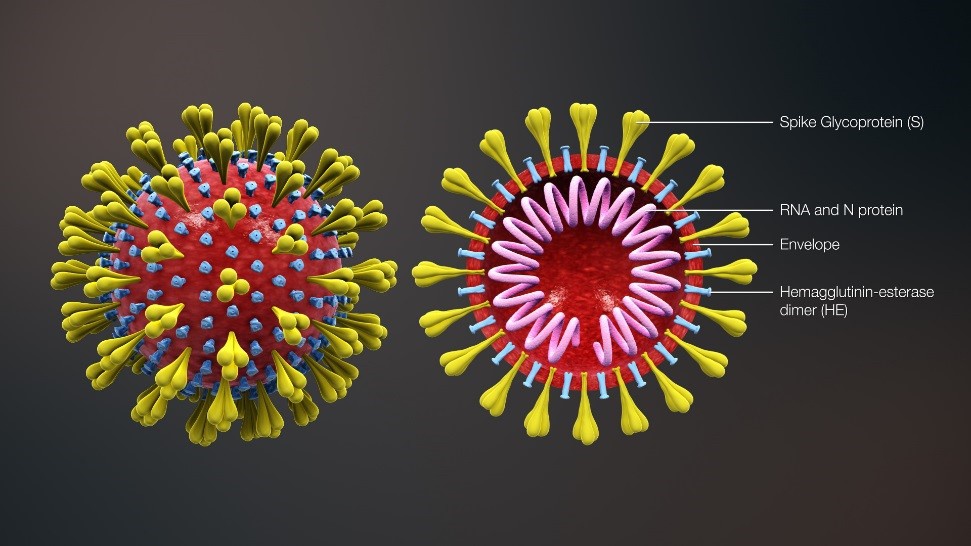
I Coronavirus si chiamano così, perché i loro bracci, le spiche (spike), visti per la prima volta al microspio elettronico, hanno dato l’impressione della presenza di una corona. Tutti i tipi sopra elencati hanno la stessa struttura di base: un filamento interno di acido nucleico Rna (associato a una proteina), che stabilisce il genoma o codice ereditario; una membrana fosfolipidica che funge da involucro; delle spiche di materiale proteico che si agganciano alle cellule da invadere. Il promemoria grafico qui sotto lo illustra per sommi capi.
Ricordiamo che l’Rna è una sequela di istruzioni che specifica quali proteine virali debbano essere sinetizzate. Se quindi cambia l’Rna cambiano quasi sempre anche le proteine; tipicamente, cambiano le proteine che costituiscono le spiche del virus.
Con questo abbiamo operato una prima scrematura, tanto per sapere di cosa parliamo. A questo punto, concentriamoci solo sul primo tipo, ovvero su Sars-CoV-2 che tanti problemi ci sta dando e che verosimilmente riguarda la domanda del quorano. Quanti ceppi esistono dunque di questo patogeno? Beh, meglio dirlo subito: la questione è molto più complessa di quanto non si pensi. Cerchiamo di semplificare.
Per rispondere bisogna innanzitutto sapere cosa possa portare un virus a modificarsi. Ebbene, la ragione risiede nelle mutazioni genetiche, cioè in singole modifiche di un nucleotide, diciamo di una “lettera”, nella catena dell’acido nucleico che compone il genoma del nostro virus. Nel caso in questione l’acido è Rna e le lettere sono A (adenina), G (guanina), C (citosina), U (uracile). All’atto di una replicazione del codice queste lettere possono essere interscambiate, soppresse o duplicate, causando la comparsa di una nuova catena di Rna modificata. Quasi sempre le mutazioni riguardano una sola lettera per volta. Ovviamente, possono essere cumulative. La mutazione della lettera U sembra dagli studi essere più frequente.
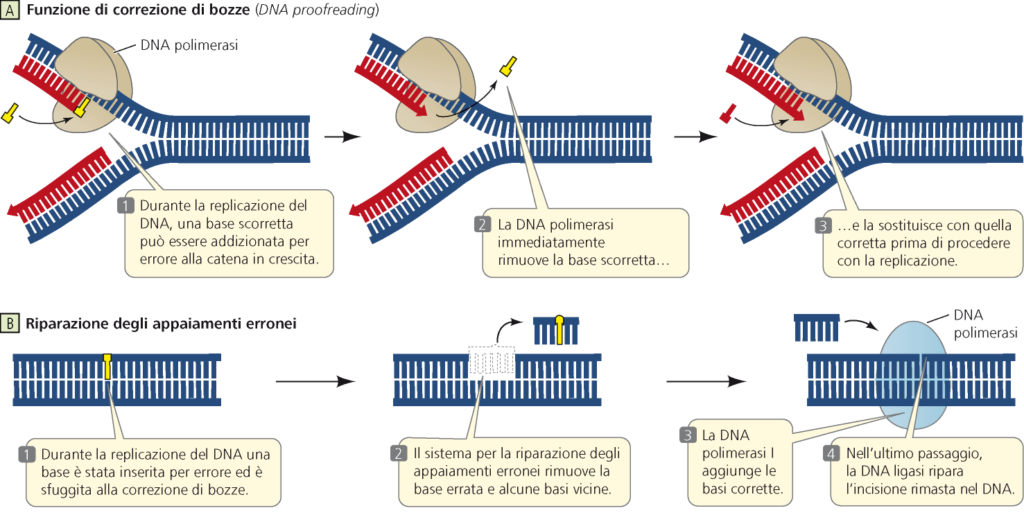
I virus a Dna possiedono di solito dei meccanismi per controllare la corretta replicazione del loro genoma; se scoprono un errore lo correggono. Si parla di sistemi di correzione delle bozze. Questi processi di controllo sono analoghi a quelli presenti nelle cellule (schema sotto). La loro presenza rende i virus a Dna più stabili nel corso delle generazioni. I virus a Rna, al contrario, raramente usufruiscono di questo controllo. Questo concorre a spiegare perché i virus dell’influenza (Influenzavirus) o il virus dell’immunodeficienza acquisita (HIV), ad esempio, mutino così rapidamente.
Il nostro Sars-CoV-2 è però uno dei virus a Rna che il meccnismo di correzione ce l’ha, seppure non sofisticato come in altri casi. Possiamo affermare subito che per questo motivo esso non si è modificato un gran che dalla sua prima comparsa a Wuhan. Questa condizione ci può allarmare, dato che implica la minaccia che questo patogeno diventi endemico, ovvero costantemente presente nella popolazione mondiale. Per versi opposti, ciò è precisamente quanto ci fa sperare su una certa durata di immunizzazione, così come la si può perseguire con una profilassi vaccinale. Ricordiamo che se il patogeno muta poco, l’immunità acquisita con un vaccino può condurre all’immunità di gregge e, poi (se non tutti sono vaccinati), all’eradicazione dell’epidemia lungo la curva discentente dei contagi.
Le mutazioni che passano alla progenie sono eventi del tutto accidentali che il sistema di correzione delle bozze si lascia sfuggire, anche questo per ragioni di casualità. Volendo assumere un ottica finalistica, potremmo dire che si tratta di “errori” che non vengono corretti, a causa di altri errori. Negli organismi viventi le mutazioni genetiche costituiscono il vero motore dell’evoluzione. Senza questi piccoli difetti la biosfera non potrebbe modificarsi nel corso del tempo; probabilmente, non potrebbe nemmeno sussistere.
I virus ben difficilmente possono considerarsi come organismi vivi (link alla mia risposta sotto), eppure anch’essi sottostanno alle leggi evolutive e quindi possono, in base alle mutazioni subite, adattarsi più o meno bene alla popolazione che infettano. Come gli organismi viventi, i virus risultano soggetti a deriva genetica e selezione naturale. Pertanto, non tutte le varianti genetiche vanno incontro allo stesso destino. Alcune, quelle più adattive, si diffondono di più, altre sono svantaggiate e tendono a scomparire. Questo in linea generale.
Va osservato che nello studio delle mutazioni di Sars-CoV-2 si procede in due modi per rilevare le varianti ereditarie: si studia il genoma a Rna e si individuano i nucleotidi alterati (le lettere) oppure si studia la catena polipeptidica (un peptide è una breve sequenza di amminoacidi) che costituisce le proteine del virus; in tal caso si individuano le alterazioni della catena polipeptidica. Infatti, una modifica della catena polinucleotidica rispecchia una mutazione nella sequenza Rna che per essa codifica.
Le mutazioni sono un fenomeno dinamico sempre in corso, quindi i differenti Sars-CoV-2 mutanti in circolazione possono differire in numero e per le loro caratteristiche al succedersi delle settimane e dei mesi. Alcuni di questi virus si estinguono, altri nuovi ne compaiono, altri ancora capita che facciano un passo indietro e si rimangino la variazione genetica; le mutazioni ad altalena (avanti e indietro) non sono infatti infrequenti tra i virus. In genere, una mutazione è dannosa e tende quindi presto a scomparire, tanto negli organismi che hanno un metabolismo, quanto nei virus.
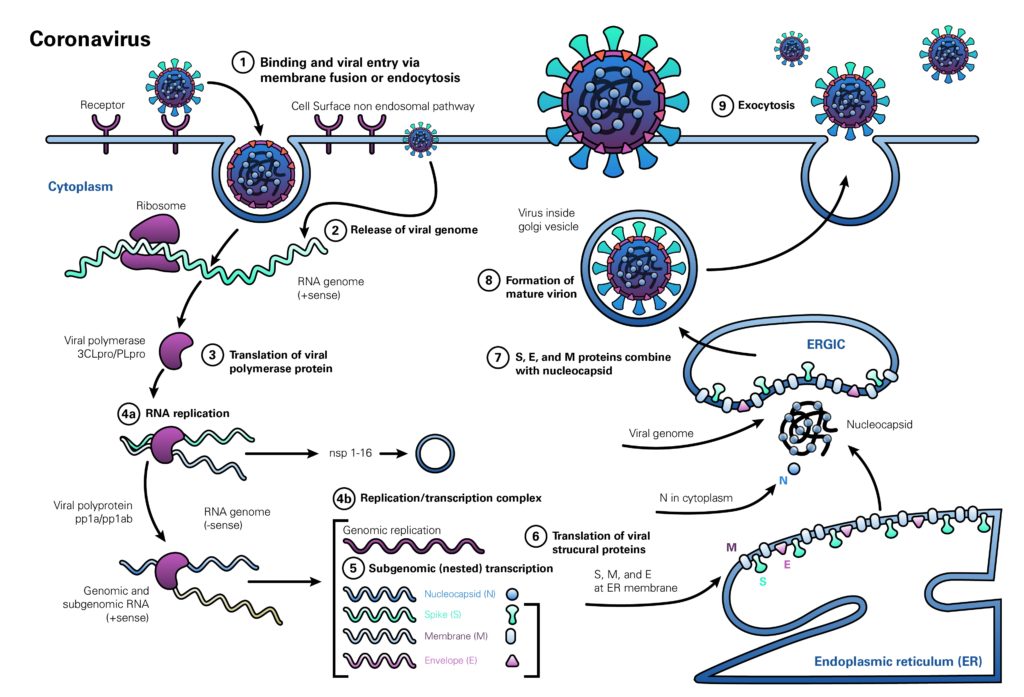
Alcune mutazioni che si stabiliscono nel genoma virale non alterano significativamente il comportamento del virus, anzi la maggior parte di tali mutazioni sono in questo senso ininfluenti. Sicuramente è così per Sars-CoV-2. Alcune mutazioni sono però più incisive. Naturalmente, tutti i ceppi virali finora identificati mantengono la stessa struttura generale e lo stesso ciclo replicativo nella cellula, così come illustra il promemoria grafico qui sotto nei dettagli del quale qui non si entra. Stiamo insomma discutendo di variazioni sul tema.
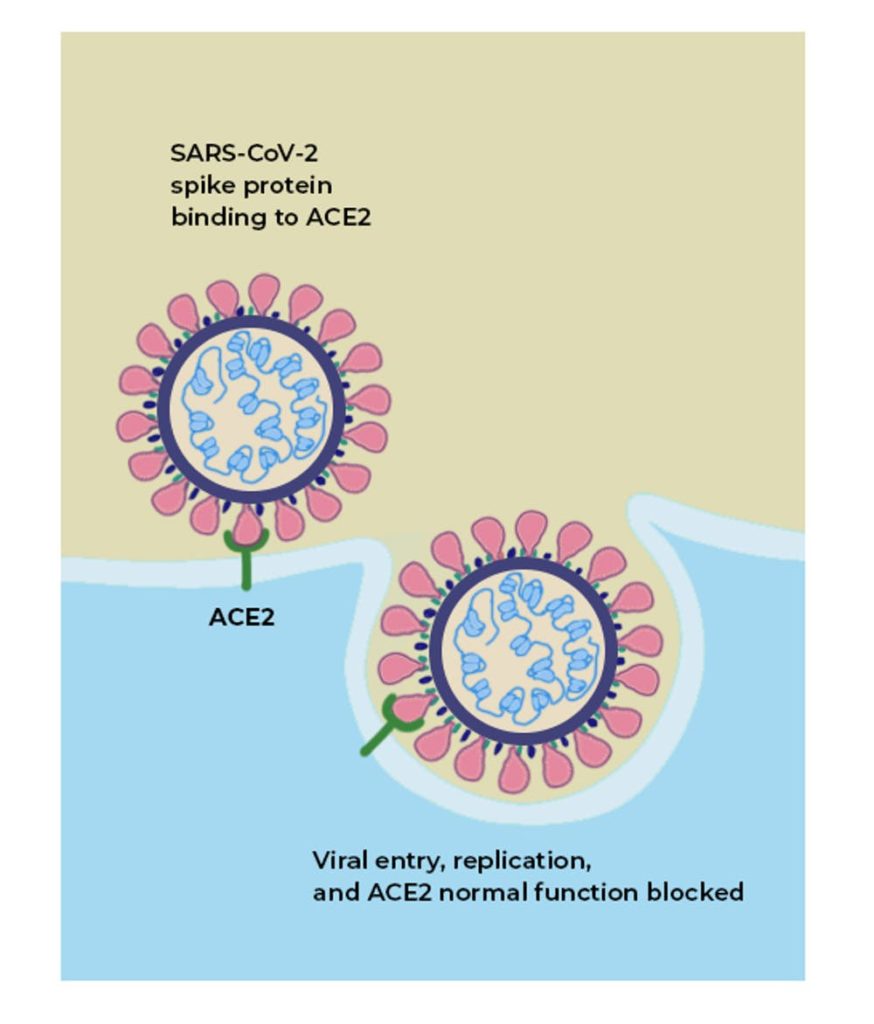
Il fatto che da un punto di vista generale siamo al cospetto di sole variazioni sul tema non deve farci dimenticare che alcune di esse possono risultare significative per quanto attiene la nostra salute o la persistenza dell’epidemia. Per esempio, è importante capire come i geni influiscano sulla dinamica d’ingresso del patogeno nelle cellule. Sappiamo che ciò succede quando il virus aggancia il recettore ACE2 presente sulla superficie della cellula aggredita. Quest’ultimo è una glicoproteina (enzima di conversione dell’angiotensina) che scatta come una serratura, quando il braccio (spica) del virus vi si connette, permettendo poi al virus intero di entrare nella cellula.
Questo processo dipende dalla forma delle spiche, più precisamente da quella delle loro estremità. Il concetto di forma è esattamente quello che riguarda anche la metafora della chiave che gira nella toppa di una serratura, permettendo di aprire una porta. Conta anche il modo in cui la chiave gira nella toppa. Nella fattispecie, conta la glicosilazione che si produce in diversi siti di contatto dell’estremità della spica con ACE2. La glicosilazione è un processo biochimico che modifica il ripiegamento delle proteine, rendendo la forma del loro sito d’interazione compatibile con l’avvio di qualche reazione biochimica. In questo caso si tratta appunto di fare scattare ACE2 sulla superficie cellulare.
La grafica semplificata qui sotto mostra come Sars-CoV-2 riesca con le sue caratteristiche spiche ad agganciare il suddetto recettore. Si nota come dopo il contatto con ACE2 la membrana cellulare subisca un’invaginazione che prelude all’ingresso del virus nella cellula. Ebbene, si capisce che tra le mutazioni più rilevanti pare ragionevole annoverare quelle che riguardano proprio questo meccanismo. Ne riparleremo più sotto.
Sars-CoV-2 muta poco, ma anch’esso non riesce a mantenersi del tutto uguale a sé stesso nel tempo. Fino ad oggi è andato incontro ad almeno alcune centinaia di mutazioni, come mostra il tracking genomico. Nemmeno tante se consideriamo che gli individui infettati nel mondo sono all’incirca mezzo miliardo (più di 50 milioni di positività riscontrate ufficialmente) e che ogni individuo contagiato può contenere miliardi di miliardi di virus. Occorre però anche prendere atto dell’impatto di quelle mutazioni. Qui subentra il discorso dei “ceppi” (strains, in inglese).
In genere, si parla di ceppi, allorquando le varianti del virus siano distinguibili sul piano biologico. Questo significa che i nucleotidi alterati (lettere nel genoma) che i virus in oggetto recano nel loro Rna conducono a reazioni differenti del sistema immunitario umano e/o determinano caratteristiche diverse di trasmissibilità del contagio. Se queste differenze non emergono in modo significativo non si raggruppano i virus in ceppi diversi.
Naturalmente, questa classificazione può risultare parzialmente imprecisa o arbitraria, ma ha comunque una sua utilità per comprendere su scala spaziale e temporale cosa stia succedendo durante un’epidemia. Attenzione, per quanto detto si capisce che il criterio di distinzione in ceppi dipende da cosa si considera come fattore discriminante. Questo spiega per quale motivo si possano leggere resoconti scientifici diversi sulla quantità di diversi ceppi in circolazione. Talvolta si parla di 20 ceppi, altre volte di 30 e altre ancora di solo 6 o 2 ceppi (per esempio, all’inizio si distingueva il tipo S dal tipo L, considerato più aggressivo).
Come si diceva, sono note diverse centinaia di mutazioni in cui l’Rna di Sars-CoV-2 è incappato dal momento della sua comparsa a Wuhan. Non possiamo però dire che siamo di fronte a un uguale numero di ceppi differenti, poiché molte di queste variazioni delle lettere del genoma non hanno alterato più di tanto le caratteristiche del virus. Due virus che si comportino in modo molto simile o uguale o non denotino differenze particolari nella loro presenza geografica rientrano nello stesso ceppo, anche se differiscono per qualche lettera (nucleotide) sui rispettivi Rna.
Estrarre dal tracking genomico (figura sotto) un raggruppamento in ceppi richiede qualche criterio che porti a una chiara e utile distinzione, altrimenti è solo un esercizio fine a sé stesso.
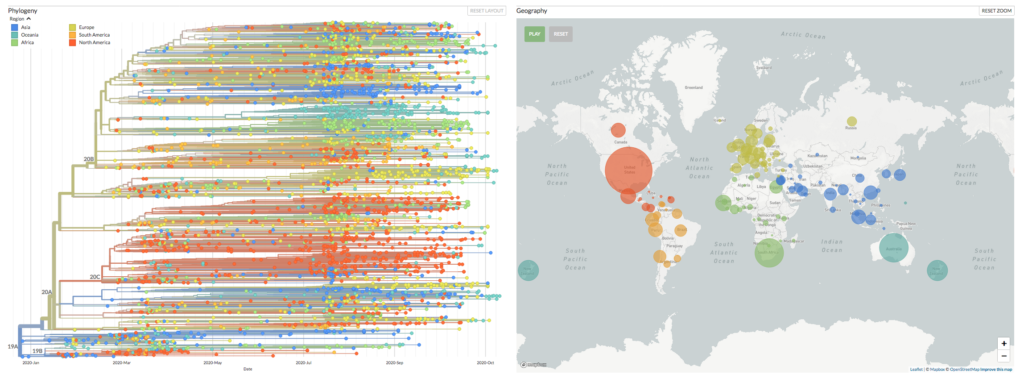
Facciamo un esempio per capire. Vedremo ora un modo di incrociare aspetti virologici con aspetti epidemiologici. Questo inquadramento è utile tanto per capire come evolva l’epidemia, quanto per individuare i criteri preventivi.
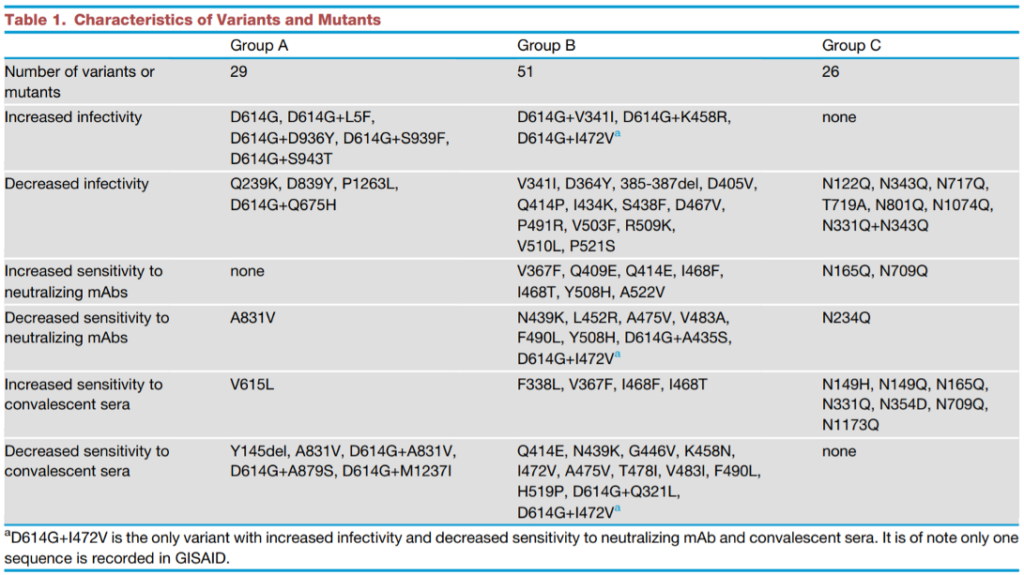
A luglio 2020 uno studio importante (pubblicato su Cell, penultimo link sotto) eseguito su 13.406 sequenze di amminoacidi delle spiche ha individuato 106 alterazioni in grado di provocare cambiamenti funzionali. Come detto, una variazione della sequenza polipeptidica riflette una mutazione genetica, cioè un cambio di lettera nell’Rna che codifica per quella stessa sequenza di amminoacidi. All’interno del campione esaminati si sono potute distinguere delle caratteristiche funzionali particolari. Il campione delle varianti genetiche è stato così suddiviso in tre gruppi virologici (A, B e C) e in 6 fattori epidemiologici. Cominciamo dai gruppi:
- Nel gruppo A sono comprese le varianti ad alta frequenza di comparsa che includano la mutazione D614G, purché non influiscano sulle caratteristiche del dominio della spica (terminazione) coinvolto nell’aggancio di ACE2.
- Nel gruppo B sono comprese le sole mutazioni che ricadono sul dominio terminale della spica, dove avviene l’interazione col recettore.
- Nel gruppo C rientrano le varianti che si differenziano per il processo di glicosilazione nei vari siti di contatto con ACE2; processo necessario per far scattare la serratura.
I fattori discriminanti che nello studio sono stati ritenuti importanti ai fini della comprensione degli effetti dell’epidemia sono stati i seguenti:
- infettività alta/bassa del virus,
- sensibilità alta/bassa del virus agli anticorpi presenti nel plasma (sera) di convalescenti,
- sensibilità alta/bassa del virus agli anticorpi monoclonali (mAbs)
Lo specchietto qui sotto illustra l’incrocio dei gruppi virologici con i fattori epidemiologici. Ogni gruppo è una colonna, mentre ogni fattore epidemiologico è indicato in una fascia orizzontale. All’interno dello specchietto si scorgono i codici alfanumerici che identificano le singole mutazioni degli amminoacidi (che riflettono mutazioni dell’Rna).
Per esempio, D614G, D614G+L5F (primo caso in tabella) individua le varianti di virus che portano la mutazione D614G da sola, ma anche quelle che recano D614G insieme alla mutazione L5F. In questo approccio tutte le varianti che appartengano a un gruppo e a un fattore epidemiologico formano un ceppo. Complessivamente, questo inquadramento derivato dall’incrocio di aspetti virologici con aspetti epidemiologici individua 15 ceppi, sulla base di 80 varianti combinate, desunte a loro volta dalla rilevazione di 106 mutazioni singole.
Come detto, le mutazioni note sono diverse centinaia. Per raggrupparle in ceppi si possono adottare vari criteri, purché utili. Per esempio, possiamo pensare di connettere le discendenze con la loro distribuzione spaziale. Abbiamo così dei raggruppamenti nelle cladi filogenetiche (diramazioni della progenie) che possiamo connettere una all’altra e alle zone geografiche che hanno conquistato sulla Terra. Questo aiuta a comprendere le modalità di diffusione del virus.
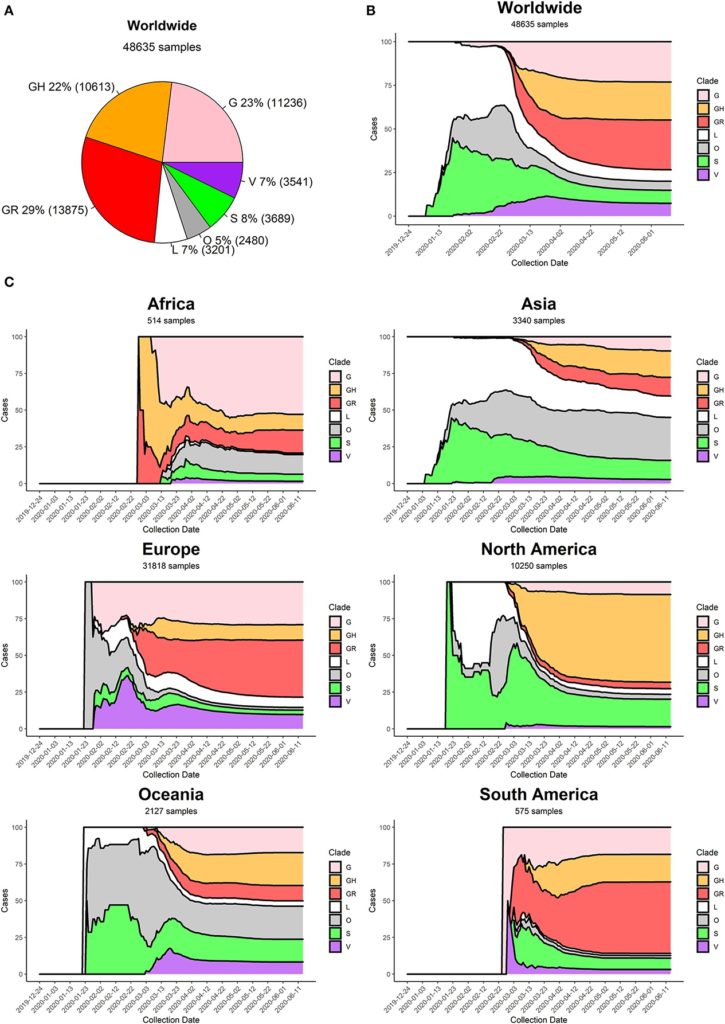
Alcuni studi, operando in questo modo, hanno identificato 6 ceppi di Sars-CoV-2. Sono così denominati: G, GH, GR, S, V e L. L’ultimo, cioè L, è il ceppo originario sorto a Wuhan che funge da riferimento per saggiare le mutazioni che sono nel frattempo sopravvenute nelle settimane e nei mesi, durante il dilagare della pandemia. Si annoverano poi altre varianti minori, più difficilmente raggruppabili e indicate con O (others).
Le immagini qui sotto raffigurano la distribuzione dei ceppi sul totale dei virus allora (luglio 2020) circolanti, nonché il relativo andamento temporale nelle differenti regioni geografiche del globo. Questo è in particolare il risultato di uno studio corposo pubblicato lo scorso luglio su Frontiers in Microbiology che ha preso in esame 48.635 genomi del virus (ultimo link).
Va osservato come in queste analisi non si distingua ormai più per la letalità delle varianti virali. Perché è così? Perché, diversamente dagli inizi (quando sembrava di poter riuscire a operare questi distinguo), col passare del tempo ci si è resi conto che gli aspetti che hanno influito sui decessi dei soggetti positivi sono troppo articolati e numerosi per poter stilare statistiche attendibili che riguardino l’aggressività intrinseca del solo virus. Sappiamo infatti quanto la letalità possa dipendere dalle norme precauzionali e dal comportamento delle persone. Ad ogni modo, sembra di poter dire che non vi siano elementi che possano chiaramente porre un ceppo come più pericoloso di un altro in termini di minaccia sulla salute del contagiato.
Stando ai dati Worldometer (non agli studi che possono variare da caso a caso), la letalità media di Sars-CoV-2 è attualmente nel mondo pari a circa 2,5% e non sembra essersi modificata. Come sappiamo, risulta differenziata in funzione dell’età in primo luogo.
Quello che è interessante osservare, giungendo con ciò dall’ultimo studio citato fino ai giorni nostri, è la prevalenza di certe varianti sul totale dei virus circolanti.
Come abbiamo visto, le istruzioni recate dal Rna codificano per la sintesi delle proteine del virus. Le spiche, cioè i bracci di Sars-CoV-2, sono tipicamente materiale proteico, quindi una variazione del Rna che le riguardi implica una modifica nella sequenza polipeptidica (cioè di amminoacidi) che compone i bracci. Di conseguenza, l’intera spica ne può risultare modificata, anche se non necessariamente ciò succede.
Ora, c’è una mutazione genetica, peraltro la prima riscontrata, che ha incontrato un enorme successo diffusivo sul globo terracqueo. Essa era già presente nell’epidemia di Wuhan e sembra avere una caratteristica vincente: consente al virus di replicarsi più rapidamente nelle cellule epiteliali del naso, da dove il contagio parte più spesso. Pertanto, quel virus mutante tende a sconfiggere le altre varianti sul piano della competizione per il contagio degli individui.
Alla fine di marzo, la mutazione compariva già nel 70% degli Rna virali in Europa e nel 60% di quelli della costa atlantica degli Usa. A maggio si riscontrava nel 95% di tutte le varianti presenti nella popolazione mondiale. Oggi, quasi tutti i virus portano quella mutazione rispetto al ceppo originario di Wuhan. Essa riguarda proprio quanto si accennava prima: la proteina che costituisce la spica di Sars-CoV-2. La variazione nucleotidica che nel Rna virale rende la proteina del braccio più efficiente è contrassegnata nel mondo della virologia come D614G. L’abbiamo già incontrata in precedenza.
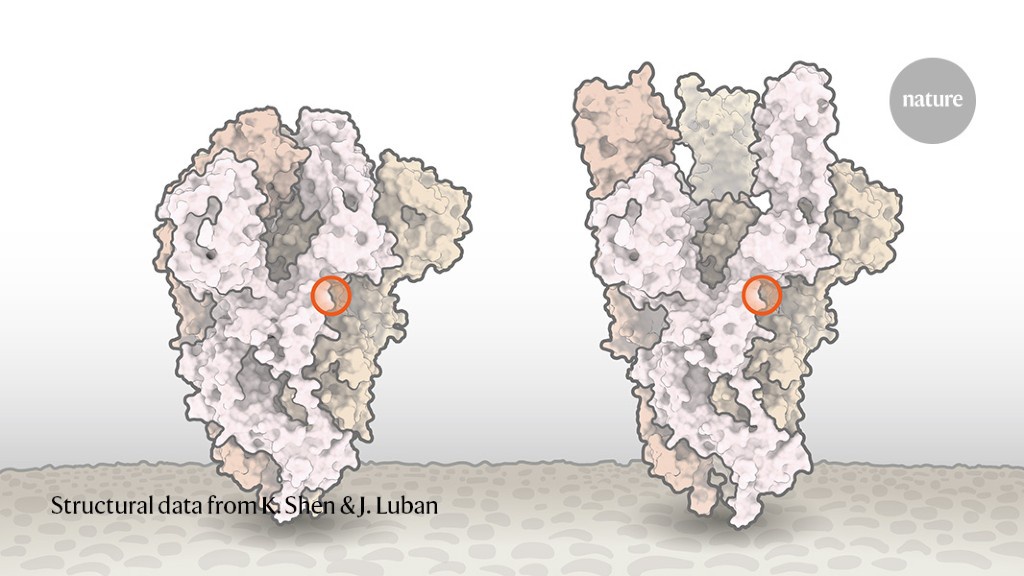
Nell’immagine qui sotto si vede la terminazione della spica proteica che è una molecola alquanto complessa, come lo sono tutte le proteine. La terminazione è la porzione più larga in alto, in basso sta la membrana del Coronavirus. Il cerchietto rosso indica la piccola porzione polipeptidica modificata dalla differente istruzione presente nel Rna del virus.
Come si nota, una piccola alterazione locale della struttura polipeptidica della proteina modifica la conformazione spaziale dell’intera molecola. Nella fattispecie, ne determina una maggiore apertura. Proprio questo consente un aggangio più efficiente al recettore ACE2 della cellula e una più veloce penetrazione della cellula stessa. Grazie al cielo, questa stessa mutazione, stabilizzatasi e ampiamente diffusasi, rende al momento il virus più facilmente “emulabile” per mezzo di un vaccino che stimoli la produzione di anticorpi specifici.
Poiché questa mutazione che riguarda la contagiosità e non incide sulla letalità è divenuta largamente maggioritaria, la distinzione in ceppi tende a perdere di significato. Certo, ci sono delle differenze genetiche e proteiche ed è sempre possibile escogitare qualche criterio di raggruppamanto. Tuttavia, non sembra al momento che sia facile tracciare demarcazioni nette che davvero tornino utili per controllare l’epidemia. In altre parole, a fronte delle mutazioni intercorse, il virus si comporta in modo molto simile in tutte le parti del mondo. Vedremo in futuro.
Per chi volesse seguire le sorti delle variazioni ereditarie di Sars-CoV-2 indico il sito Nextstrain che raccoglie i dati inerenti la distribuzione delle varie cladi virali sulla Terra e nel corso del tempo. A questo database di tracking genomico (connesso con Gisaid), organicamente elaborato e rappresentato in modo grafico molto completo, si riferiscono praticamente tutti i ricercatori del mondo. L’approccio non è immediato, occorre studiarsi un po’ la cosa, ma la fonte è davvero preziosa.
Accesso generale alla pagina Home: Nextstrain
Accesso alla distribuzione cladica di Sars-CoV-2: auspice
Se volete approfondire l’argomento sulla storia di Virus e Batteri, fate riferimento a un bellissimo articolo sul sito amico:

Fonti:
What is coronavirus? The different types of coronaviruses
Risposta di Roberto Weitnauer a I virus sono esseri viventi o non viventi? E perché?
SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo
The coronavirus is mutating – does it matter?
Spike mutation D614G alters SARS-CoV-2 fitness
The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity
Geographic and Genomic Distribution of SARS-CoV-2 Mutations