Studio mostra picchi proteici sviluppati in laboratorio coerenti con il virus COVID-19
coronavirus mutations
Un nuovo studio internazionale ha scoperto che le proprietà chiave dei picchi del virus SARS-CoV-2 che causa il COVID-19 sono coerenti con quelle di diversi picchi proteici sviluppati in laboratorio, progettati per imitare il virus infettivo.
Una componente centrale nella progettazione di test sierologici e vaccini per la protezione contro il COVID-19 è la produzione di “picchi” proteici. Questi picchi ricombinanti imitano da vicino quelli che sporgono dalla superficie del virus infettivo e attivano il sistema immunitario del corpo.
Le punte prodotte in laboratorio vengono utilizzate anche per i test sierologici (noti anche come test sugli anticorpi) e come reagenti di ricerca. I risultati mostrano come il picco virale prodotto con metodi diversi nei laboratori di tutto il mondo sia molto simile e rassicura sul fatto che il picco può essere prodotto in modo robusto con variazioni minime tra i laboratori.
I picchi del virus SARS-CoV-2 sono ricoperti di zuccheri, noti come glicani, che usano per camuffarsi dal sistema immunitario umano. L’abbondanza di questi glicani ha il potenziale per creare discrepanze significative tra gli studi che utilizzano diversi picchi ricombinanti.
In questo nuovo studio, pubblicato sulla rivista Biochemistry , il team di ricerca ha studiato i rivestimenti di glicani sugli spike ricombinanti sviluppati in cinque laboratori in tutto il mondo e li ha confrontati con quelli sugli spike del virus infettivo.
“La velocità con cui la comunità scientifica si è mossa per affrontare la pandemia di COVID-19 ha esercitato una notevole pressione sui laboratori di tutto il mondo per convalidare rapidamente i loro risultati”, ha spiegato Max Crispin, professore di glicobiologia all’Università di Southampton, che ha guidato lo studio. “Nell’ultimo anno abbiamo assistito allo sviluppo di vaccini in tutto il mondo a un ritmo senza precedenti e il rapido sviluppo e la convalida delle proteine ricombinanti sono stati fondamentali per questa storia di successo”, ha continuato.
Nell’aprile 2020, il professor Crispin e il suo team dell’Università di Southampton hanno mappato per la prima volta il rivestimento di glicani del picco di SARS-CoV-2. Nel presente studio, estendono la loro analisi per esaminare il picco ricombinante sviluppato nei laboratori dell’Amsterdam University Medical Centre, della Harvard Medical School, dell’Università di Oxford e della società svizzera ExcellGene. È stato dimostrato che tutti i diversi lotti di proteine spike imitano le caratteristiche chiave della glicosilazione dei virioni inattivati analizzati alla Tsinghua University, in Cina.
Lo studio ha anche utilizzato metodi computazionali per esaminare le caratteristiche proteiche che stavano plasmando alcune delle caratteristiche di glicosilazione osservate in tutti i campioni. Il Dr. Peter Bond, ricercatore principale senior presso il Bioinformatics Institute of the Agency for Science, Technology and Research (A*STAR), Singapore, che ha guidato il lavoro computazionale, ha dichiarato: “Il nostro modello ci ha permesso di far luce su come la proteina influenza il struttura dei glicani e perché la glicosilazione era così coerente. Questo approccio predittivo potrebbe anche essere di potenziale valore nello sviluppo di terapie contro nuove varianti o altri virus emergenti”.
“La capacità di produrre imitazioni della proteina spike SARS-CoV-2 con alta fedeltà in molti laboratori diversi, che ricapitolano tutte le firme glicani del virus autentico , è di notevole beneficio per la progettazione del vaccino, i test sugli anticorpi e la scoperta di farmaci”, ha concluso il professor Crispin.
Lo studio, “Controllo sterico sito-specifico della glicosilazione del picco SARS-CoV-2”, è stato pubblicato su Biochemistry .
Astratto
Un principio centrale nella progettazione dei vaccini è la visualizzazione di antigeni nativi nell’induzione dell’immunità protettiva. L’abbondanza di glicani legati all’N attraverso la proteina spike di SARS-CoV-2 è una potenziale fonte di eterogeneità tra i molti diversi candidati al vaccino in esame. Qui, indaghiamo la glicosilazione delle proteine spike ricombinanti SARS-CoV-2 provenienti da cinque diversi laboratori e le confrontiamo con la proteina S del virus infettivo, coltivata in cellule Vero. Troviamo modelli che vengono conservati in tutti i campioni e questo può essere associato allo stallo specifico del sito della maturazione del glicano che funge da reporter altamente sensibile della struttura proteica. Le simulazioni di dinamica molecolare di un picco completamente glicosilato supportano un modello di restrizioni steriche che modellano l’elaborazione enzimatica dei glicani.Questi risultati suggeriscono che la glicosilazione immunogena SARS-CoV-2 basata su spike ricombinante ricapitola in modo riproducibile le firme della glicosilazione virale.
La pandemia di Coronavirus 2019 (COVID-19) ha portato allo sviluppo di una serie senza precedenti di vaccini candidati contro il patogeno causale, la sindrome respiratoria acuta grave-coronavirus-2 (SARS-CoV-2). Tutti gli approcci mirano a fornire le caratteristiche molecolari del virus per indurre l’immunità. La glicoproteina virale spike, chiamata anche proteina S, è emersa come l’obiettivo principale degli sforzi di progettazione del vaccino poiché gli anticorpi contro questo bersaglio possono offrire un’immunità robusta. (1-6) In modo incoraggiante, la neutralizzazione può facilmente verificarsi nonostante l’ampia gamma di glicani legati all’N distribuiti attraverso il picco virale coerente con numerose vulnerabilità in questo cosiddetto scudo di glicani. (7) Nonostante queste osservazioni, la glicosilazione è emersa come un parametro importante nello sviluppo di vaccini per SARS-CoV-1 e SARS-CoV-2. (8,9) Lo stato di elaborazione della glicosilazione può influenzare il traffico di immunogeni nel sistema linfatico, (10) influenzare la presentazione di epitopi criptici sia nativi che indesiderati, (11)e rivelano la misura in cui gli immunogeni ricapitolano l’architettura virale nativa. Sta emergendo anche la prova che la glicosilazione può influenzare in qualche modo l’interazione tra SARS-CoV-2 e il suo recettore bersaglio, l’enzima di conversione dell’angiotensina 2 (ACE2). (12-15)Ogni protomero della proteina trimerica SARS-CoV-2 S contiene almeno 22 sequoni di glicosilazione legati a N che dirigono l’attacco dei glicani dell’ospite a specifici residui di Asn. Questa estesa glicosilazione è importante nel ripiegamento proteico mediato dalla lectina e nella stabilizzazione diretta del ripiegamento proteico. (16) Inoltre, alcuni glicani maturano in modo incompleto durante la biogenesi e possono portare alla presentazione di glicani immaturi che terminano con residui di mannosio che possono agire come ligandi per il riconoscimento immunitario innato. (17-19)
Nonostante il focus sulla proteina S negli sforzi di sviluppo del vaccino, c’è stata una notevole divergenza nei meccanismi di consegna. In un approccio, un acido nucleico che codifica lo spike viene consegnato tramite mRNA o con un vettore virale. (4−6,20−23)La proteina S risultante viene assemblata e glicosilata dal tessuto ospite. In un approccio contrastante, la proteina S può essere prodotta in modo ricombinante come proteina ricombinante utilizzando linee cellulari di mammiferi o insetti o utilizzando approcci basati su virus inattivati che consentono una caratterizzazione dettagliata dell’immunogeno prima della consegna. (24-28) La glicosilazione dell’immunogeno può essere influenzata da fattori specifici delle condizioni di produzione come il tipo di cellula o le condizioni di coltura cellulare; (29,30)tuttavia, anche la progettazione costruttiva e l’architettura delle proteine possono avere un impatto sostanziale. Ad esempio, siti oligomannosio sottoprocessati possono verificarsi in siti stericamente nascosti agli enzimi mannosidasi dell’ospite dall’architettura terziaria o quaternaria, compreso l’offuscamento da parte della struttura proteica e del glicano adiacenti. (18,31)Gli immunogeni che mostrano un’architettura nativa ricapitolano questi siti di glicosilazione dell’oligomannosio. Al contrario, il design dell’immunogeno può avere un impatto negativo sulla presentazione della glicosilazione nativa. È importante sottolineare che, nonostante le differenze nella biosintesi della proteina S nei virioni e nei sistemi di espressione dei mammiferi, sembrano generare glicosilazione sostanzialmente simile. (32)Tuttavia, il successo di un’ampia gamma di diverse piattaforme di vaccini che mostrano una diversa glicosilazione della proteina S indica che la glicosilazione di tipo nativo non è un prerequisito per un vaccino di successo. Nonostante questa osservazione, la comprensione della glicosilazione della proteina S aiuterà a confrontare il materiale impiegato in diversi studi sierologici e sui vaccini e aiuterà a definire l’impatto di queste ampie caratteristiche della superficie della proteina.
La natura flessibile ed eterogenea della glicosilazione legata all’N necessita di metodologie ausiliarie oltre alla microscopia crioelettronica o alla cristallografia a raggi X per caratterizzare questa parte chiave della struttura della proteina S. L’analisi dei glicani sito-specifica che impiega la cromatografia liquida-spettrometria di massa è un approccio ampiamente utilizzato per ottenere queste informazioni. (33-37)Con il progredire della ricerca sulla struttura e sulla funzione della proteina SARS-CoV-2 S, sono diventati evidenti maggiori dettagli sullo scudo glicano della proteina S. Le analisi della proteina S trimerica ricombinante hanno rivelato la glicosilazione legata all’N divergente dalle glicoproteine dell’ospite in presenza di glicani di tipo oligomannosio sottoelaborati in diversi siti. (7,15,38) Analisi comparative con proteine S monomeriche e trimeriche hanno rivelato differenze sito-specifiche nella glicosilazione per quanto riguarda sia i glicani di tipo oligomannosio che la presentazione dell’acido sialico. (38) L’analisi della proteina S da cellule di insetto ha dimostrato che i glicani di tipo oligomannosio sono stati conservati sulla proteina S trimerica, in particolare a N234. (39) Inoltre, studi di dinamica molecolare (MD) hanno proposto che il sito N234 svolga un ruolo nella stabilizzazione del dominio di legame del recettore (RBD) in una conformazione esposta “up”. (31)La presenza di glicani di tipo oligomannosio sottoprocessati più grandi, come Man 9 GlcNAc 2 , sia sulla proteina S derivata dai mammiferi che da quella degli insetti fornisce un’indicazione che la struttura della proteina S sta guidando la presentazione di questi glicani. Studi successivi hanno studiato la presentazione di glicani legati all’N sulla proteina S prodotta per la vaccinazione, in particolare la proteina S a lunghezza intera Novavax e la proteina S isolate in seguito alla somministrazione del vettore ChAdOx-nCoV-19. (39,40)Le firme dei glicani osservate erano ampiamente in accordo con le analisi precedenti. Tuttavia, questi studi hanno coinvolto il troncamento delle strutture glicaniche utilizzando il trattamento con glicosidasi, che è utile per classificare i glicani in glicani ad alto mannosio o di tipo complesso e determinare la potenziale occupazione del sito di N-glicosilazione (PNGS) su piccole quantità di materiale ma non consente per l’identificazione di cambiamenti nell’elaborazione del glicano terminale come la sialilazione. Per il RBD monomerico è stata studiata anche l’elaborazione dei glicani dei due siti glicani legati all’N situati sull’RBD. Questi siti presentano alti livelli di glicani di tipo complesso, (41,42) e poiché la maggior parte degli anticorpi generati contro la proteina SARS-CoV-2 S mira all’RBD, è importante caratterizzare completamente la struttura dell’RBD, inclusa la presentazione dei glicani.
I glicani N-Linked sono altamente dinamici e possono variare sostanzialmente nella composizione chimica all’interno di un singolo lotto di proteine. Sebbene la glicosilazione sia eterogenea, è importante per la progettazione terapeutica e vaccinale capire se glicoformi simili sorgono su diverse piattaforme di espressione proteica da fonti diverse per garantire che la superficie antigenica della proteina S rimanga coerente quando viene utilizzata come immunogeno o nei test sierologici. (43-46) Ciò è particolarmente importante in quanto l’elaborazione del glicano può essere influenzata da conformazioni proteiche avverse. (47)Qui, descriviamo un approccio integrato che include la cromatografia liquida-spettrometria di massa (LC-MS) e simulazioni MD per comprendere le caratteristiche di glicosilazione attraverso proteine S ricombinanti da diverse fonti. Esploriamo quindi la misura in cui queste proteine ricombinanti riproducono la glicosilazione della proteina SARS-CoV-2 S di origine virale prodotta da cellule Vero coltivate che è stata ottenuta da uno studio precedente. (32)Abbiamo impiegato un approccio analitico identico per determinare la composizione del glicano in ciascun sito, illustrando la conservazione attraverso la proteina ricombinante e il materiale derivato dal virione. Inoltre, abbiamo analizzato la glicosilazione della proteina ricombinante monomerica RBD, poiché è stata precedentemente esplorata come vaccino a subunità e candidato per i test sierologici. (48,49)Abbiamo quindi confrontato l’analisi del glicano sito-specifica dei due siti situati nel RBD di SARS-CoV-2, N331 e N343, tra il monomero e il trimero e abbiamo rivelato un ampio consenso nell’elaborazione del glicano, con qualche modesto cambiamento nell’elaborazione di glicani di tipo complesso. Abbiamo anche eseguito un’analisi comparativa sul MERS-CoV RBD. Ciò contrasta con l’elaborazione del glicano del monomero RBD SARS-CoV-2 rispetto alla proteina S, con i siti di glicani MERS-CoV RBD trimerici che presentano un’elaborazione del glicano ristretta, probabilmente a causa del mascheramento conformazionale di questi siti, sia da parte dei glicani prossimali che da proteine vicine scontri, sulla proteina trimerica MERS-CoV S. (50,51)Abbiamo ulteriormente combinato i dati glicani specifici del sito della proteina SARS-CoV-2 S con simulazioni MD. Queste simulazioni rivelano gradi distinti di accessibilità tra diversi siti di glicani attraverso la proteina che sono ampiamente correlati ai loro stati di elaborazione. Presi insieme, i nostri risultati rivelano i siti N-glicani strutturali conservati nella proteina S rispetto al picco virale nativo, che guida la somiglianza nella glicosilazione tra le proteine S da fonti e metodi di produzione disparati. Comprendere la glicosilazione della proteina S aiuterà nell’analisi del vaccino e nel lavoro sierologico della risposta globale al COVID-19.
Purificazione delle proteine SARS-CoV-2 S (Harvard)
Per esprimere un ectodominio stabilizzato della proteina spike, un gene sintetico che codifica i residui 1–1208 di SARS-CoV-2 Spike con il sito di scissione della furina (residui 682–685) sostituito da una sequenza “GGSG”, sostituzioni di prolina ai residui 986 e 987 , e un motivo di trimerizzazione foldon seguito da un tag six-His C-terminale è stato creato e clonato nel vettore di espressione di mammifero pCMV-IRES-puro (Codex BioSolutions, Inc., Gaithersburg, MD). Il costrutto di espressione è stato transfettato in modo transitorio in cellule HEK 293T utilizzando polietilenimina (Polysciences, Inc., Warrington, PA). La proteina è stata purificata dai surnatanti cellulari utilizzando la resina Ni-NTA (Qiagen) e le frazioni eluite contenenti la proteina S sono state raggruppate, concentrate e ulteriormente purificate mediante cromatografia di filtrazione su gel su una colonna Superose 6 (GE Healthcare).
Purificazione delle proteine SARS-CoV-2 S (Oxford)
Il dominio extracellulare della proteina spike SARS-CoV-2 è stato clonato nel vettore pHLsec (52)e comprendeva i residui M1–Q1208 (UniProtKB P0DTC2 ) con mutazioni R682G/R683S/R685S (sequenza di riconoscimento della furina) e K986P/V987P, seguito dalla regione di trimerizzazione della fibritina, un sito di scissione della proteasi HRV3C, un tag di otto His e un doppio- Etichetta dello streptococco al C-terminale. (28,53)L’ectodominio spike è stato espresso mediante trasfezione transitoria di cellule HEK293T (ATCC, CRL-3216) per 6 giorni a 30 ° C. Il mezzo condizionato è stato dializzato contro tampone salino 2× tamponato con fosfato (pH 7,4) (Sigma-Aldrich). L’ectodominio dello spike è stato purificato utilizzando la resina Strep-Tactin Superflow (IBA Lifesciences) seguita da cromatografia di esclusione dimensionale utilizzando una colonna Superose 6 Aumenta (GE Healthcare) equilibrata in 200 mM NaCl, 2 mM Tris-HCl (pH 8,0) e 0,02% NaN 3 a 21°C.
Purificazione delle proteine SARS-CoV-2 S (Amsterdam)
Il costrutto proteico SARS-CoV-2 S è stato progettato ed espresso come descritto in precedenza. (1)In breve, un gene SARS-CoV-2 S che codifica per i residui 1–1138 (WuhanHu-1; voce GenBank MN908947.3 ) è stato ordinato (Genscript) e clonato in un plasmide pPPI4 contenente un dominio di trimerizzazione T4 seguito da un tag esaistidina di Pst I – BamaHI digestione e legatura. Le modifiche consistono nel sostituire le posizioni degli amminoacidi 986 e 987 con proline e rimuovere il sito di scissione della furina sostituendo gli amminoacidi 682-685 con glicine. La proteina S è stata espressa nelle cellule HEK 293F. Circa 1,0 milioni di cellule/mL, mantenute in terreno Freestyle (Gibco), sono state trasfettate aggiungendo una miscela di PEImax (1 μg/μL) e plasmide SARS-CoV-2 S (312,5 μg/L) in un rapporto 3:1 in OptiMEM. Dopo 6 giorni, i surnatanti sono stati centrifugati per 30 minuti a 4000 rpm e filtrati utilizzando un filtro Steritop (Merck Millipore, 0,22 μm). La proteina S è stata purificata dal surnatante mediante purificazione per affinità utilizzando perline di agarosio Ni-NTA e gli eluati sono stati concentrati e il tampone è stato scambiato con PBS utilizzando filtri Vivaspin con un cutoff di peso molecolare di 100 kDa (GE Healthcare).La concentrazione della proteina S è stata determinata con il metodo Nanodrop utilizzando il peso molecolare peptidico delle proteine e il coefficiente di estinzione come determinato dal software online ExPASy (ProtParam).
Produzione e purificazione di proteine del trimero di spike SARS-CoV-2 da cellule CHO
Per la produzione basata su CHO di una proteina spike trimerica, è stato utilizzato il costrutto Spike_ΔCter_ΔFurin_2P_T4_His. (54)In breve, sulla base del vettore CHO pXLG-6 (ExcellGene SA), contenente un marker di resistenza alla puromicina ed elementi di espressione ottimizzati, è stata inserita la sequenza spike variante, contenente una sequenza del sito di clivaggio della furina scambled, una sequenza a due proline per stabilizzare il proteina nella forma di prefusione e un DNA di trimerizzazione 3′ T4, seguito da una sequenza di tag esaistidina. Dalla trasfezione chimica (CHO4Tx, ExcellGene) alla selezione clonale e all’isolamento di linee cellulari produttrici di proteine fino alla successiva espansione delle cellule e alla produzione, tutti i passaggi sono stati eseguiti in una coltura in sospensione in un mezzo chimicamente definito con stretta aderenza alle raccomandazioni normative.
La produzione è stata eseguita in contenitori agitati orbitalmente (TubeSpin bioreactor 600, TPP, Trasadingen, Svizzera) o in palloni agitatori tipo Erlenmeyer da 5 L in un incubatore Kuhner Shaker ISF4-X (Adolf Kühner AG, Birsfelden, Svizzera), impostato a 37 °C , 5% CO 2 , controllo dell’umidità e agitazione a 150 rpm con raggio di spostamento di 50 mm. È stato implementato un processo fed-batch fino a quando i surnatanti non sono stati raccolti il giorno 10. La vitalità della coltura cellulare, raggiungendo ∼25 × 10 6cellule/mL, è rimasto nell’intervallo 90-100% fino al raccolto. I fluidi di coltura di produzione sono stati sottoposti a purificazione mediante cromatografia di affinità dopo filtrazione di profondità per rimuovere le cellule. Il caricamento, il lavaggio e l’eluizione da una colonna Ni-Sepharose (Cytiva) sono stati ottimizzati, seguendo i suggerimenti dei produttori di resina. Il flusso di prodotto eluito è stato caricato su una colonna di esclusione dimensionale (SEC, Superdex 200 pg, Cytiva) per ulteriore purificazione, a seguito di una fase di concentrazione tramite filtrazione a flusso tangenziale.
Costruzione ed espressione del plasmide delle proteine del dominio di legame del recettore
Per esprimere i domini di legame del recettore (RBD) di SARS-CoV-2 (residui 320-527), SARS-CoV-1 (residui 307-513) e MERS-CoV (residui 368-587), abbiamo amplificato i frammenti di DNA mediante reazione a catena della polimerasi utilizzando i plasmidi ectodomain della proteina SARS-CoV-2, SARS-CoV-1 e MERS-CoV S come modelli. I frammenti di DNA sono stati clonati nel vettore phCMV3 (Genlantis) in cornice con la sequenza leader dell’attivatore del plasminogeno tissutale (TPA). Per facilitare la purificazione e la biotinilazione della proteina, abbiamo fuso la proteina con un tag Six-His C-terminale e AviTag distanziati da linker GS.
I costrutti RBD sono stati espressi mediante trasfezione transitoria di cellule FreeStyle293F (Thermo Fisher). Per una trasfezione di 1 L, abbiamo aggiunto 350 μg dei plasmidi codificanti RBD a 16 mL di Transfectagro (Corning) e 1,5 mL di 40K PEI (1 mg/mL) in 16 mL di Transfectagro in provette coniche da 50 mL separate. I mezzi contenenti i plasmidi sono stati filtrati con un’unità filtrante sottovuoto monouso Steriflip da 0,22 μm (Millipore Sigma) e miscelati delicatamente con PEI. La miscela plasmide/PEI è stata incubata per 30 minuti e aggiunta delicatamente a 1 L di cellule ad una concentrazione di 1 milione di cellule/mL. I surnatanti di coltura sono stati raccolti dopo 4 giorni e caricati su una colonna di resina HisPur Ni-NTA (Thermo Fisher). Dopo un lavaggio con imidazolo 25 mM, la proteina dalla colonna è stata eluita con imidazolo 250 mM.L’eluato è stato scambiato tampone con PBS e concentrato utilizzando provette Amicon 10K. Le proteine RBD sono state ulteriormente purificate tramite cromatografia ad esclusione dimensionale utilizzando una colonna Superdex 200 Incremento 10/300 GL (GE Healthcare).
Preparazione e analisi del campione mediante LC-MS
I dati della spettrometria di massa per la glicosilazione derivata dal virione sono stati ottenuti utilizzando i protocolli descritti da Yao et al. (32)e rianalizzato. Per la proteina S ricombinante e l’RBD, aliquote di 30 μg di proteine SARS-CoV-2 S sono state denaturate per 1 ora in 50 mM Tris-HCl (pH 8,0) contenente 6 M di urea e 5 mM di ditiotreitolo (DTT). Successivamente, le proteine S sono state ridotte e alchilate aggiungendo 20 mM di IAA e incubate per 1 ora al buio, seguita da incubazione con DTT per eliminare qualsiasi residuo di IAA. Le proteine S alchilate sono state scambiate con tampone in 50 mM Tris-HCl (pH 8,0) utilizzando colonne Vivaspin (3 kDa) e digerite separatamente durante la notte utilizzando tripsina, chimotripsina (grado spettrometria di massa, Promega) o proteasi α-litica (Sigma-Aldrich ) con un rapporto di 1:30 (p/p). I peptidi sono stati essiccati ed estratti utilizzando un C18 Zip-tip (Merck Millipore). I peptidi sono stati nuovamente essiccati, risospesi in acido formico 0,1%,e analizzato da nanoLC-ESI MS con un sistema Easy-nLC 1200 (Thermo Fisher Scientific) accoppiato a uno spettrometro di massa Orbitrap Fusion (Thermo Fisher Scientific) utilizzando la frammentazione di dissociazione indotta da collisione (HCD) ad alta energia. I peptidi sono stati separati utilizzando una colonna EasySpray PepMap RSLC C18 (75 μm × 75 cm). Una colonna di intrappolamento [PepMap 100 C18 3 μm (dimensione delle particelle), 75 μm × 2 cm] è stata utilizzata in linea con il cromatografo liquido prima della separazione con la colonna analitica. Le condizioni LC erano le seguenti: gradiente lineare di 275 min costituito dallo 0% al 32% di acetonitrile in acido formico allo 0,1% in 240 min seguito da 35 min di acetonitrile all’80% in acido formico allo 0,1%. La portata è stata fissata a 300 nL/min. La tensione dello spray è stata impostata su 2,7 kV e la temperatura del capillare riscaldato è stata impostata su 40 °C. La temperatura del tubo di trasferimento ionico è stata impostata a 275 ° C.Il raggio di scansione era m / z 400–1600. L’energia di collisione dell’HCD è stata impostata al 50%. Il rilevamento del precursore e il rilevamento del frammento sono stati eseguiti utilizzando Orbitrap con una risoluzione MS1 di 100000 e una risoluzione MS2 di 30000. Il target AGC per un MS1 di 4 × 10 5 e un MS2 di 5 × 10 4 e tempi di iniezione di 50 ms per MS1 e 54 ms di MS2.
Elaborazione dati di dati LC-MS
I dati sulla frammentazione del glicopeptide sono stati estratti dal file raw Byos (versione 3.5, Protein Metrics Inc.). I seguenti parametri sono stati utilizzati per le ricerche di dati in Byonic. La tolleranza di massa del precursore è stata fissata a 4 e 10 ppm per i frammenti. Le modifiche peptidiche incluse nella ricerca includono Cys carbamidomethyl, Metossidazione, Glu → pyroGlu, Gln → pyroGln e N deammidazione. Per ogni digest di proteasi, è stato utilizzato un nodo di ricerca separato con parametri di digestione appropriati per ciascuna proteasi (tripsina RK, chimotripsina YFW e ALP TASV) utilizzando una ricerca semispecifica con due scissioni mancate. È stato applicato un tasso di falsa scoperta (FDR) dell’1%. Tutti e tre i digest sono stati combinati in un unico file per l’analisi a valle. Sono stati sommati tutti gli stati di carica per un singolo glicopeptide.I dati di frammentazione del glicopeptide sono stati valutati manualmente per ciascun glicopeptide; il peptide è stato valutato come vero positivo quando sono stati osservati gli ioni del frammento b e y corretti insieme agli ioni ossonio corrispondenti al glicano identificato. La metrica della proteina 309 della libreria di N-glicani di mammifero è stata modificata per includere glicani solfatati e specie di mannosio fosforilato, sebbene non siano stati rilevati glicani di mannosio fosforilato su nessuno dei campioni analizzati. Le quantità relative (determinate confrontando il XIC di ciascun glicopeptide, sommando gli stati di carica) di ciascun glicano in ciascun sito, nonché la proporzione non occupata, sono state determinate confrontando le aree cromatografiche estratte per i diversi glicotipi con quelle di una sequenza peptidica identica. I glicani sono stati classificati in base alla composizione rilevata. HexNAc(2),Hex(9–3) è stato classificato da M9 a M3. Ognuna di queste composizioni che è stata rilevata con un fucosio è classificata come FM. HexNAc(3)Hex(5–6)NeuAc(0–1) è stato classificato come ibrido con HexNAc(3)Hex(5–6)Fuc(1)NeuAc(0–1) classificato come Fhybrid. I glicani di tipo complesso sono stati classificati in base al numero di antenne processate e allo stato di fucosilazione. I glicani complessi sono classificati come HexNAc(3)(X), HexNAc(3)(F)(X), HexNAc(4)(X), HexNAc(4)(F)(X), HexNAc(5)(X) , HexNAc(5)(F)(X), HexNAc(6+)(X) e HexNAc(6+)(F)(X). I glicani principali sono qualsiasi glicano più piccolo di HexNAc(2)Hex(3). I glicani sono stati ulteriormente classificati in base al loro stato di elaborazione. Un glicano era classificato come fucosilato o sialilato se conteneva almeno un residuo di fucosio o acido sialico.I glicani agalattosilati consistevano di composizioni nelle categorie sopra menzionate per glicani di tipo complesso che possedevano solo tre residui di esosi per glicani di tipo complesso, o se la composizione era un glicano di tipo ibrido, allora questo veniva aumentato a cinque esosi. Se il glicano di tipo complesso non conteneva acido sialico ma più di tre esosi per glicani di tipo complesso o cinque esosi per glicani di tipo ibrido, allora veniva conteggiato come galattosilato. I glicani solfatati sono stati inclusi nelle categorie sopra descritte, quindi un glicano può essere sia nelle categorie galattosilati che solfatati, per esempio.Se il glicano di tipo complesso non conteneva acido sialico ma più di tre esosi per glicani di tipo complesso o cinque esosi per glicani di tipo ibrido, allora veniva conteggiato come galattosilato. I glicani solfatati sono stati inclusi nelle categorie sopra descritte, quindi un glicano può essere sia nelle categorie galattosilati che solfatati, per esempio.Se il glicano di tipo complesso non conteneva acido sialico ma più di tre esosi per glicani di tipo complesso o cinque esosi per glicani di tipo ibrido, allora veniva conteggiato come galattosilato. I glicani solfatati sono stati inclusi nelle categorie sopra descritte, quindi un glicano può essere sia nelle categorie galattosilati che solfatati, per esempio.
Modellazione integrativa e simulazione di dinamica molecolare
Il modello della proteina S è stato costruito utilizzando Modeler 9.21 (55)con tre modelli strutturali: (i) la struttura cryo-EM di SARS-CoV-2 S ECD nello stato aperto [file 6VSB della banca dati proteica (PDB) ], (28)(ii) la struttura NMR del dominio SAR-CoV S HR2 (file PDB 2FXP ), (56)e (iii) la struttura NMR del dominio HIV-1 gp-41 TM (file PDB 5JYN ). (57)I loop mancanti nel dominio N-terminale e nel dominio C-terminale dell’ECD sono stati modellati utilizzando la struttura crio-EM dell’ECD S nello stato chiuso, che è stata risolta a una risoluzione più elevata (file PDB 6XR8 ). (58) Sono stati costruiti un totale di 10 modelli e tre modelli migliori (basati sul punteggio energetico proteico ottimizzato discreto più basso (59)) sono stati sottoposti a un’ulteriore valutazione stereochimica mediante l’analisi di Ramachandran. (60)È stato quindi selezionato il modello con il minor numero di outlier. I glicani Man-9 sono stati aggiunti a tutti i 22 siti di glicosilazione utilizzando il server web CHARMM-GUI Glycan Reader e Modeler. (61) Tre residui di cisteina sul dominio C-terminale (C1236, C1240 e C1243) sono stati palmitoilati poiché hanno dimostrato di essere importanti nella fusione cellulare. (62) Il modello di proteina S glicosilata è stato quindi incorporato in una membrana modello ER/Golgi Intermedio Complex (ERGIC) costruita utilizzando CHARMM-GUI Membrane Builder, (63) che conteneva il 47% di fosfatidilcolina (PC), il 20% di fosfatidiletanolammina (PE), l’11% di fosfatidilinositolo fosfato (PIP), il 7% di fosfatidilserina (PS) e il 15% di colesterolo. (64-66)
Il sistema è stato parametrizzato utilizzando il campo di forza CHARMM36. (67)Le molecole d’acqua TIP3P sono state utilizzate per solvatare il sistema e sono stati aggiunti 0,15 M di sale NaCl per neutralizzarlo. Il sistema è stato quindi sottoposto a minimizzazione ed equilibrazione graduale dell’energia con vincoli di posizione e diedri decrescenti, seguendo il protocollo standard CHARMM-GUI. (68) Una simulazione di produzione di 200 ns è stata condotta con la temperatura mantenuta a 310 K utilizzando il termostato Nosé-Hoover. (69,70) La pressione è stata mantenuta a 1 atm utilizzando un accoppiamento semi-isotropo al barostato di Parrinello-Rahman. (71) L’elettrostatica è stata calcolata utilizzando il metodo Ewald della rete di particelle lisce (72)con un cutoff nello spazio reale di 1.2 nm, mentre le interazioni di van der Waals sono state troncate a 1.2 nm con una funzione di livellamento dell’interruttore di forza applicata tra 1.0 e 1.2 nm. L’algoritmo LINCS è stato utilizzato per vincolare tutti i legami covalenti che coinvolgono atomi di idrogeno, (73)ed è stato impiegato un passo temporale di integrazione di 2 fs. Tutte le simulazioni sono state eseguite utilizzando GROMACS 2018 (74) e visualizzato in VMD. (75)Le analisi dell’area di superficie accessibile (ASA) e del contatto glicano sono state eseguite utilizzando gli strumenti GROMACS integrati gmx sasa e gmx select .
Espressione e purificazione della proteina S ricombinante da fonti multiple
Per definire la variabilità nella glicosilazione della proteina S ricombinante trimerica e confrontare la proteina S ricombinante e derivata da virus, abbiamo ottenuto preparazioni di proteina S ricombinante da una serie di laboratori. Questi includono l’Amsterdam Medical Center (Amsterdam), la Harvard Medical School (Harvard), la Svizzera, il Wellcome Center for Human Genetics (Oxford) e le scienze biologiche dell’Università del Texas ad Austin (Southampton/Texas). (1,28,53,54,76,77)Sebbene ci siano piccole differenze tra i costrutti utilizzati per produrre la proteina S, il design generale è simile. Ciò comporta un troncamento della proteina S prima del C-terminale, sostituzione del sito di scissione della furina tra S1 e S2 con un linker “GSAS” o altrimenti mutato, un motivo di trimerizzazione della fibritina T4 C-terminale e stabilizzazione della prefusione S conformazioni proteiche mediante sostituzioni di prolina. (28,50,78)Le proteine ricombinanti analizzate contengono 22 siti glicani legati all’N ad eccezione della preparazione “Amsterdam” che è stata troncata prima degli ultimi tre potenziali siti di glicosilazione legati all’N. È stato eseguito un allineamento delle proteine ricombinanti, e i siti glicani sono numerati secondo la numerazione usata in precedenti rapporti della glicosilazione sito-specifica della proteina S stabilizzata con 2P ( Figura SI ). (7)Tutte le proteine ricombinanti sono state espresse in cellule renali embrionali umane 293 (HEK293) con le preparazioni proteiche di Amsterdam, Harvard e Southampton/Texas prodotte nelle cellule HEK293F e la preparazione proteica Oxford S prodotta nelle cellule HEK293T. La proteina S “Svizzera” è stata espressa nelle cellule dell’ovaio di criceto cinese (CHO). Dopo l’espressione e la purificazione, è stato utilizzato un approccio identico per preparare i campioni ricombinanti per l’analisi mediante LC-MS, coinvolgendo tre digeriti di proteasi separati (tripsina, chimotripsina e proteasi α-litica). Dopo l’analisi LC-MS, la glicosilazione sito-specifica di questi campioni ricombinanti è stata confrontata con quella della proteina S derivata da virus precedentemente pubblicata che abbiamo cercato utilizzando gli stessi parametri analitici utilizzati per le proteine ricombinanti. (32)
Conservazione di glicani sottoprocessati su proteina S ricombinante trimerica e derivata viralmente
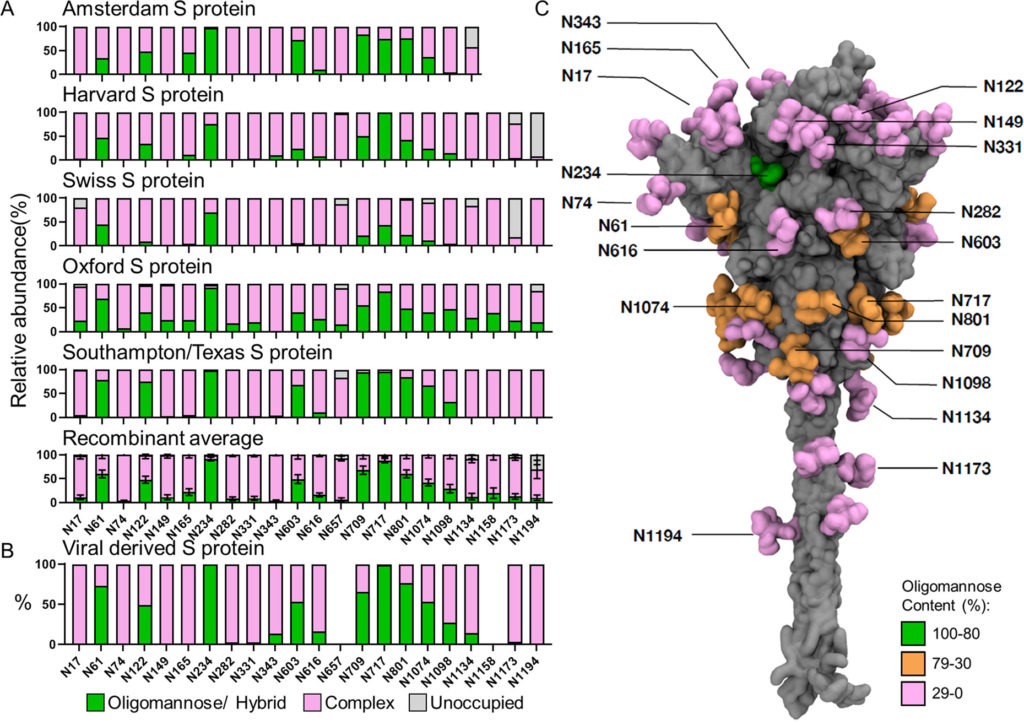
I glicani di tipo oligomannosio e di tipo ibrido sottoprocessati sono comuni sulle glicoproteine virali e si verificano comunemente a causa di scontri sterici mediati da glicani o proteine con ER e enzimi mannosidasi residenti nel Golgi, che terminano il percorso di elaborazione del glicano. Poiché la presenza di questi glicani è legata alla struttura quaternaria della proteina, cambiamenti nell’abbondanza di queste glicoforme possono indicare cambiamenti nella struttura fine della glicoproteina. Per studiare l’abbondanza di questi glicani, abbiamo semplificato le composizioni eterogenee di glicani rilevate da LC-MS in tre categorie: (1) glicani di tipo oligomannosio e di tipo ibrido, (2) glicani di tipo complesso e (3) la proporzione di PNGS privi di un glicano legato all’N. Complessivamente,i campioni ricombinanti hanno ricapitolato l’elaborazione del glicano osservata sulla proteina S di origine virale ( Figura 1 A, B). I siti sul materiale derivato da virus con un’abbondanza di glicani sottoelaborati > 30% includono N61, N122, N234, N603, N709, N717, N801 e N1074 ( Figura 1 B). Con poche eccezioni, tutti i campioni ricombinanti analizzati presentano anche almeno il 30% di glicani di tipo oligomannosio in questi siti ( Tabella S1 ).
Biochemistry 2021
Figura 1. Glicosilazione sito-specifica della proteina S ricombinante e derivata da virus da più laboratori. (A) Analisi glicani sito-specifica della proteina S ricombinante espressa e purificata in luoghi diversi. I grafici a barre rappresentano le proporzioni relative di glicoformi presenti in ciascun sito, inclusa la proporzione di PNGS che non sono state modificate da un glicano legato all’N. Le proporzioni di glicani di tipo oligomannoso e ibrido sono colorate in verde. I glicani di tipo complesso elaborati sono colorati in rosa e le proporzioni dei siti non occupati sono colorate in grigio. L’istituto che ha fornito la proteina S per l’analisi è elencato sopra ogni grafico. I dati Texas/Southampton sono riprodotti da Chawla et al. (dati non pubblicati). Le composizioni medie della proteina S ricombinante sono state calcolate utilizzando tutti i campioni.Le barre rappresentano la media ± l’errore standard della media di tutti i campioni ricombinanti analizzati. (B) L’analisi sito-specifica derivata viralmente è stata eseguita utilizzando i dati acquisiti da Yao et al. e classificati nello stesso modo sopra descritto.(32)I dati per i siti N657 e N1158 non possono essere ottenuti e non sono rappresentati. (C) Modello a lunghezza intera che mostra la glicosilazione oligomannosio sito-specifica della proteina S derivata dal virus. Una descrizione di come è stato generato questo modello è disponibile in Materiali e metodi . Sia le proteine che i glicani sono mostrati nella rappresentazione superficiale; il primo è colorato in grigio e il secondo colorato in base al contenuto di oligomannosio come mostrato nella tabella S1 (verde per l’80-100%, arancione per il 30-79% e rosa per lo 0-29%).
Per confrontare la variabilità della glicosilazione di tipo oligomannosio e ibrido tra tutte le proteine ricombinanti, le composizioni sono state mediate e visualizzate con l’errore standard della media (SEM) delle preparazioni di proteine S di diversi laboratori ( Figura 1A,B). Questa analisi ha rivelato un ampio consenso sull’elaborazione dei glicani per quanto riguarda i glicani ad alto contenuto di mannosio, con variazioni localizzate. Un esempio di notevole omogeneità è il sito N234, che presenta alti livelli di glicani sottoprocessati in tutti i campioni analizzati. Questa elaborazione è stata conservata sulla proteina S di origine virale. Il materiale ricombinante possedeva bassi livelli di glicani di tipo oligomannosio in diversi siti attraverso la proteina non presenti sul materiale di origine virale. Ad esempio, il materiale analizzato da Oxford aveva almeno il 5% di glicani di tipo oligomannosio in ogni sito tranne N343 ( Figura 1UN). Questo moderato aumento globale del livello di glicani di tipo oligomannosio potrebbe potenzialmente derivare da due fonti; il primo è che i preparati ricombinanti utilizzano mutazioni stabilizzanti che potrebbero generare una struttura più compatta che limita la capacità di agire degli enzimi di elaborazione del glicano, e questo è supportato dall’osservazione che la proteina S derivata viralmente mostra conformazioni metastabili nel NTD, probabilmente aumentando l’accessibilità a particolari siti glicani. (32) Il secondo potrebbe essere che la maggiore resa di proteine ottenuta durante l’espressione della proteina ricombinante pone limitazioni alla capacità della cellula di elaborare il gran numero di siti glicani presenti sulla proteina S.
L’occupazione di glicani è un altro parametro importante da considerare per la progettazione dell’immunogeno. È stato dimostrato che la sottooccupazione di PNGS glicoproteici ricombinanti rispetto a quella delle loro controparti virali provoca la presentazione di epitopi distraenti non neutralizzanti ed è importante da monitorare. (79,80)Come nel caso dell’HIV-1 Env di origine virale, la proteina SARS-CoV-2 S di origine virale ha mostrato alti livelli di occupazione di glicani in tutti i siti analizzati ( Figura 1 B). Dopo il confronto di questi dati con i dati di glicosilazione specifici del sito di consenso, la maggior parte dei PNGS sulla proteina S ricombinante è altamente occupata. Le eccezioni a questo sono al C-terminale, dove diversi campioni analizzati mostrano una ridotta occupazione di glicani indicata da una popolazione più ampia di siti non occupati ( Figura 1UN). Questi siti che mostrano proporzioni maggiori di glicani non occupati al C-terminale sono probabilmente dovuti al troncamento delle proteine ricombinanti richieste per la solubilizzazione; è stato precedentemente riportato che la vicinanza di un sequone glicano al C-terminale può influenzare la sua occupazione. (81)
La modellazione della glicosilazione sito-specifica di materiale di origine virale consente di contestualizzare spazialmente in tre dimensioni la glicosilazione della proteina S. Utilizzando la struttura crio-EM della proteina S ECD allo stato aperto (un RBD nella “conformazione up” e due RBD nella “conformazione down”) (file PDB 6VSB ), (28)così come le strutture NMR del dominio SARS-CoV HR2 (file PDB 2FXP ) (56)e il dominio HIV-1 gp-41 TM (file PDB 5JYN ), (57)è stato generato un modello completo della proteina S (dettagli in Materiali e Metodi ). Questo modello include siti di glicosilazione che spesso non vengono risolti in quanto presenti su anse variabili o lungo la regione del gambo flessibile della proteina S. Questo modello ha dimostrato la diffusione di diversi stati di elaborazione del glicano attraverso la proteina S ( Figura 1C). Come con altri modelli pubblicati di glicosilazione spike, il sito N234 è ampiamente sepolto all’interno della superficie proteica prossimale all’RBD e razionalizza la presentazione conservata di glicani di tipo oligomannosio su materiale ricombinante e derivato viralmente poiché gli enzimi di elaborazione del glicano non sono in grado di elaborare completamente questi siti. I siti glicani situati su anse variabili e sul gambo esposto della proteina S sono molto più elaborati poiché questi siti sono più esposti agli enzimi di elaborazione del glicano.
Elaborazione di glicani divergenti in siti che presentano glicani di tipo complesso
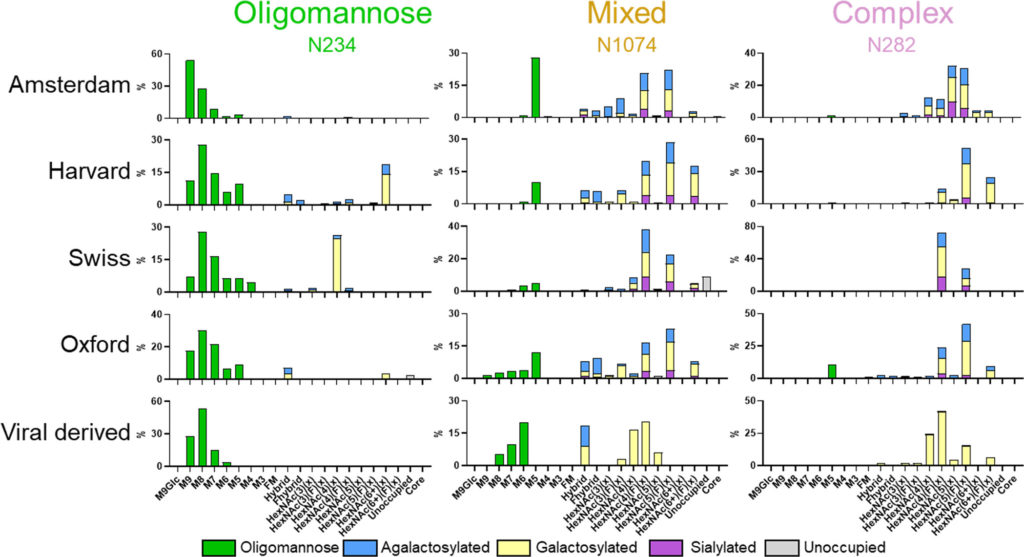
Mentre la sottoelaborazione dei glicani può fornire informazioni relative alla struttura quaternaria della glicoproteina, la maggior parte dei glicani attraverso la proteina S può essere elaborata oltre queste glicoforme. I siti glicani non sottoposti alle stesse pressioni strutturali, come in siti ristretti come N234, saranno processati in modo analogo a quelli presentati sulla maggior parte delle glicoproteine dell’ospite. Confrontando le composizioni di glicani sito-specifiche nei siti di esempio, abbiamo cercato di comprendere la variabilità nell’elaborazione del glicano tra più preparazioni di proteina S. Per questa analisi, abbiamo confrontato i dati di glicosilazione della proteina ricombinante presentati in questo studio (Amsterdam, Harvard, Svizzera,e Oxford) con i dati per la proteina S derivata da virus precedentemente riportati ma analizzati utilizzando gli stessi parametri di ricerca utilizzati per le proteine ricombinanti. Per facilitare il confronto, abbiamo selezionato un sito che presentava livelli elevati di glicani di tipo oligomannosio (N234), uno che presentava una miscela di stati di elaborazione (N1074) e un sito popolato da glicani di tipo complesso (N282) ( Figura 2 ). Il confronto della glicosilazione sito-specifica di N234 ha dimostrato la conservazione dell’elaborazione del glicano di tipo oligomannosio. Su quattro dei cinque campioni presentati in Figura 2 , il glicano più abbondante rilevato era Man 8 GlcNAc 2 e sulla proteina Amsterdam S predominava Man 9 GlcNAc 2 , che è marginalmente meno elaborato. I campioni di Harvard e Swiss hanno mostrato livelli elevati di elaborazione di glicani a N234, ma questi glicani erano abbondanti solo ∼ 20% ed erano ancora i siti meno elaborati su questi campioni ( Figura 2e Tabella S1 ).
biochemistry 2021
Figura 2. Confronto di composizioni dettagliate tra siti con stati di elaborazione differenziali. Composizioni di glicani a N234, N1074 e N282. Per tutti i campioni analizzati, i glicani sono stati classificati e colorati in base alle composizioni rilevate. I glicani di tipo oligomannoso (M9-M4) sono colorati in verde. I glicani di tipo ibrido, quelli contenenti tre HexNAc e almeno cinque esosi, sono stati colorati come per i glicani di tipo complesso perché un braccio può essere processato in modo simile. I glicani di tipo complesso sono stati classificati in base al numero di residui di HexNAc rilevati e alla presenza o assenza di fucosio. I glicani centrali rappresentano qualsiasi composizione rilevata più piccola di HexNAc 2 Hex3. Per i glicani ibridi e di tipo complesso, le barre sono colorate per rappresentare l’elaborazione terminale presente. Il blu rappresenta agalattosilato, giallo galattosilato (contenente almeno un galattosio) e viola sialilato (contenente almeno un acido sialico). La proporzione di PNG non occupati è colorata in grigio.
Il sito N1074 presentava una miscela diversificata di glicani di tipo oligomannosio, ibridi e complessi in tutti i campioni analizzati. L’abbondanza di glicani di tipo oligomannosio era più variabile in questo sito con il contenuto totale di glicani di tipo oligomannosio variabile dall’11% al 35% ( Figura 2). La composizione predominante a N1074 variava da campione a campione. Per la preparazione ricombinante di Amsterdam e la proteina S di origine virale, un glicano di tipo oligomannosio era il glicano più abbondante a N1074; tuttavia, per gli altri campioni, un glicano completamente processato era più abbondante. Per tutti i campioni analizzati, N1074 presentava diversi stati di elaborazione del glicano. L’elaborazione di glicani di tipo complesso con diversi monosaccaridi può influenzare la funzione della glicoproteina a cui sono legati; per esempio, la sialilazione può prolungare l’emivita di una glicoproteina nel corpo. (82)Man mano che progrediscono attraverso l’apparato di Golgi, i glicani possono essere elaborati mediante l’aggiunta di fucosio, galattosio e acido sialico. L’abbondanza di questi monosaccaridi è influenzata dalla cellula da cui sono prodotti e dalle condizioni di coltura o dalle composizioni del terreno in cui vengono prodotte le proteine ricombinanti. Tipicamente, le glicoproteine prodotte dalle cellule HEK293F e CHO presentano glicani di tipo complesso con alti livelli di fucosilazione e galattosilazione ma livelli inferiori di sialilazione. (83,84)Il trattamento di N1074 e N282 lo dimostra con la maggior parte dei glicani di tipo complesso recanti almeno un fucosio su tutte le preparazioni S ricombinanti. Allo stesso modo, la maggior parte dei glicani è galattosilata, sebbene siano presenti popolazioni di glicani prive di qualsiasi elaborazione oltre la ramificazione della N- acetilglucosamina. I glicani di tipo ibrido, in cui un braccio del glicano viene elaborato ed elaborato come per i glicani di tipo complesso e uno rimane sottoprocessato, sono presenti in abbondanze inferiori attraverso N1074 nelle preparazioni ricombinanti. Questi glicani di tipo ibrido sono anche generalmente di scarsa abbondanza sulle glicoproteine dei mammiferi. (85)
Mentre le composizioni di glicani di tipo complesso e ibrido sono variabili, ci sono alcune tendenze visibili quando si confronta l’elaborazione del glicano tra la proteina S ricombinante e quella derivata dal virus. La differenza più netta è la mancanza di residui di acido sialico non solo su N1074 e N282 ma anche su tutti i PNGS di materiale derivato da virus ( Figura 2 e Tabella SI ). Facendo la media di tutte le proteine S prodotte in modo ricombinante e confrontandole con le proteine S derivate da virus, troviamo una diminuzione di 21 punti percentuali (pp) nella sialilazione ( Tabella S2). Anche la fucosilazione del materiale derivato dal virus è inferiore sia su N1074 che su N282, e questa tendenza è nuovamente rispecchiata in tutti i siti, con la proteina S derivata da virus che possiede una diminuzione di 16 pp nella fucosilazione ( Tabella S2 ). La differenza finale è nella ramificazione del glicano. Il numero di antenne glicani processate può essere dedotto dal numero rilevato di N-acetilesosammina (HexNAc) determinati mediante LC-MS. Più HexNAc è presente su una composizione di glicani, più il glicano tende ad essere ramificato; per esempio, HexNAc(6) può corrispondere a un glicano tetra-antennario, mentre HexNAc(4) è biantennario. Poiché l’elaborazione del glicano è eterogenea, questo cambiamento è più sottile. Tuttavia, i glicani di tipo complesso di N1074 e N282 sono meno ramificati sulla proteina S virale rispetto alle proteine S ricombinanti, con una diminuzione di 30 pp nell’abbondanza di HexNac(5) e HexNAc(6) corrispondente a tri- e tetra -glicani antennari ( Figura 2 ). Questi cambiamenti sono evidenti anche su altri siti elaborati come N165 e N1158 ( Tabella S2 ).
Questi cambiamenti contrastano con analisi simili che confrontano Env derivato da virus e varianti ricombinanti solubili. Per HIV-1 Env, si osserva un aumento della ramificazione dei glicani e della sialilazione nei siti che presentano glicani di tipo complesso. (86) Queste modifiche erano presenti anche su Env. (87)Diversi fattori potrebbero influenzare questi cambiamenti. Il primo è che il legame di membrana offerto all’Env virale porta i glicani in prossimità degli enzimi di elaborazione del glicano, e il secondo è che la cellula produttrice aveva livelli di espressione maggiori degli enzimi glicosiltransferasi coinvolti nell’elaborazione del glicano. La proteina SARS-CoV-2 S in un contesto virale è probabilmente soggetta a vincoli simili; tuttavia, l’elaborazione del glicano è distinta. Un fattore che potrebbe essere importante è il germogliamento precoce del virione SARS-CoV-2 nel complesso intermedio ER/Golgi (ERGIC) che può allontanare la proteina spike dalle glicosiltransferasi presenti nel trans-Golgi rispetto a Env, che rimane attaccato a membrana, prossimale alle glicosiltransferasi. (88)La scelta della cellula produttrice e delle condizioni di coltura del virus, in questo caso le cellule Vero, che sono derivate dalle cellule renali di Cercopithecus aethiops (scimmia verde africana), può anche influenzare l’elaborazione del glicano poiché i livelli di espressione degli enzimi glicosiltransferasi possono spiegare per l’attaccamento indebolito dell’acido sialico e del fucosio osservato sulla proteina S virale rispetto alla proteina S ricombinante. Precedenti studi hanno evidenziato la bassa abbondanza di porzioni di acido sialico sulle cellule Vero; (89)tuttavia, questi cambiamenti potrebbero verificarsi anche a causa delle condizioni di coltura utilizzate per la produzione virale. Poiché questi cambiamenti nella glicosilazione probabilmente non influiscono sull’immunogenicità dei mimetici degli spike virali, come evidenziato dall’elevata efficacia di diversi vaccini, queste osservazioni rimangono importanti quando si considera come il virus possa interagire con il sistema immunitario tramite interazioni con la lectina e possono essere informative quando si considerano test antigenici e purificazioni utilizzando reagenti leganti glicani. Nonostante le differenze osservate, notiamo che la glicosilazione sito-specifica del materiale derivato dal virus qui delineato è coerente con l’analisi del glicano precedentemente riportata dalla proteina S derivata dal virus prodotta nelle cellule epiteliali polmonari Calu-3. (90)
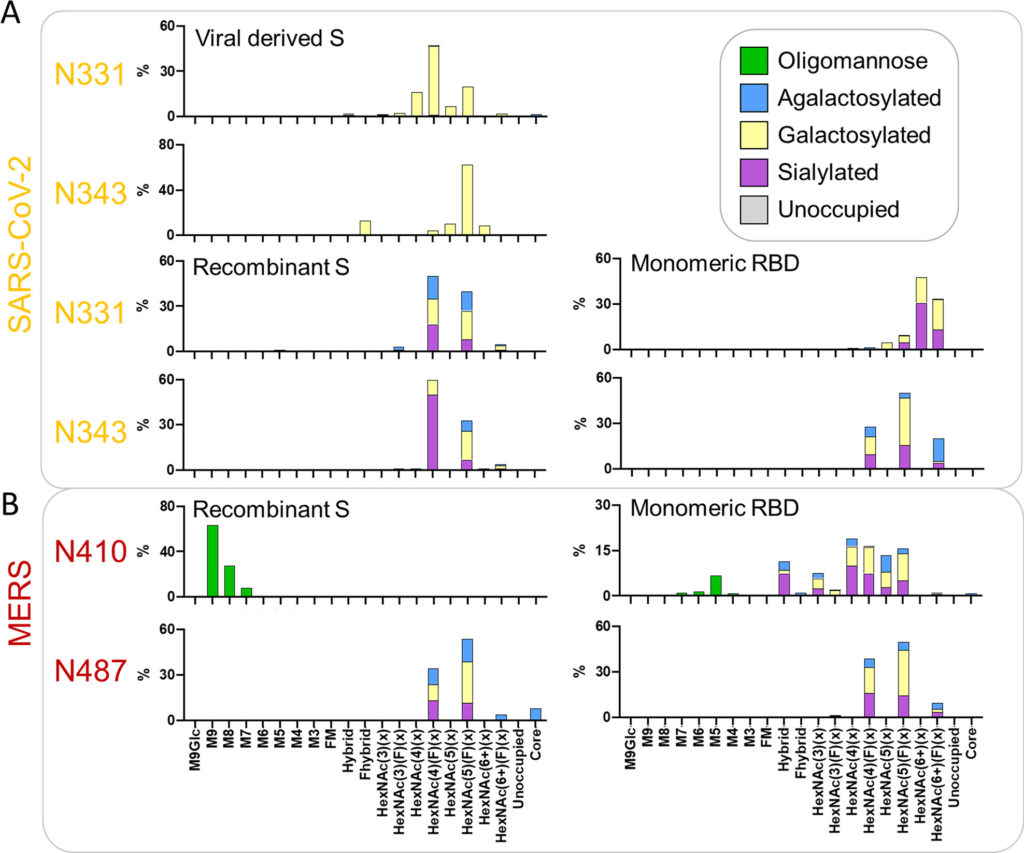
L’espressione dei costrutti RBD monomerici influisce sull’elaborazione del glicano
La nostra analisi dimostra che la glicosilazione della proteina S è influenzata dall’architettura della proteina quaternaria e da altri fattori. Successivamente abbiamo cercato di confrontare la glicosilazione dell’RBD monomerico ricombinante solubile con quella della proteina S trimerica ricombinante e derivata da virus. Abbiamo espresso e purificato l’RBD ricombinante e confrontato la glicosilazione sito-specifica dei due siti glicani situati nell’RBD, N331 e N343, con quelli osservati sulla proteina S ricombinante e sulla proteina S derivata dal virus riportati in precedenza. (7,32)Nel complesso, i glicani N331 e N343 in tutti i formati di espressione sono stati altamente elaborati, con glicani di tipo oligomannosio rilevati da pochi a nessun ( Figura 3 A, B e Tabella S3 ). Per i siti RBD presentati sulla proteina S derivata viralmente, i glicani di tipo complesso osservati erano simili a quelli osservati su N282 con la maggior parte dei PNGS occupati da glicani bi- e triantennari [HexNAc(4) e HexNAc(5)] ( Figura 3UN). La maggior parte di questi glicani era fucosilata; tuttavia, una grande proporzione dei glicani di tipo complesso su S di origine virale mancava di fucosilazione (24% N331 e 20% N343). Come con altri siti sulla proteina S di origine virale, sono stati osservati livelli minimi di sialilazione e la maggioranza dei glicani possedeva almeno un residuo di galattosio.
Biochemistry 2021
Figura 3. Analisi comparativa della glicosilazione dei due PNGS sull’RBD per la proteina S derivata viralmente, la proteina S ricombinante e l’RBD monomerico. (A) Composizioni di glicani sito-specifiche dettagliate dei due siti situati nel RBD di SARS-CoV-2. I dati sulla proteina S ricombinante sono riprodotti da Chawla et al. (dati non pubblicati) e i dati per i siti RBD della proteina S virale sono stati ottenuti da ref(32). I dati sul glicano sito-specifici sono presentati come delineato nella Figura 2 . (B) Composizioni sito-specifiche per i siti N-glicani situati nel RBD di MERS-CoV quando espressi come parte di una proteina S ricombinante solubile rispetto al solo RBD. I dati per la proteina MERS-CoV S sono stati ottenuti da ref(51). I dati sul glicano sito-specifici sono presentati come delineato nella Figura 2 .
In confronto, i due siti RBD della proteina S solubile ricombinante sono altamente fucosilati, con quasi il 100% dei glicani a N331 e N343 contenenti almeno un fucosio ( Figura 3 B). Come con la proteina S di origine virale, la maggior parte dei glicani sono glicani di tipo complesso bi- e triantennario. I siti RBD della proteina S ricombinante sono anche più sialilati di N331 e N343 sulla proteina S di derivazione virale, con il 28% di glicani N331 contenenti almeno un acido sialico e il 60% di quelli su N343 ( Tabella S3). È interessante notare che quando l’RBD è espresso come monomero, ci sono ulteriori sottili cambiamenti rispetto alla proteina S ricombinante e derivata viralmente. Il cambiamento più importante è nella ramificazione del glicano; considerando che i siti glicani RBD della proteina S trimerica ricombinante e derivata viralmente possiedono piccole quantità di glicani tetra-antennari, sull’RBD monomerico circa un terzo dei glicani in N331 e un quinto dei glicani in N343 sono costituiti da queste strutture ramificate più grandi ( Figura 3). Rispetto alla proteina S ricombinante, i siti RBD monomerici possiedono anche livelli inferiori di glicani biantennari. Nonostante questi cambiamenti, sia la proteina S trimerica ricombinante che i siti RBD monomerici hanno alti livelli di fucosilazione. Questi risultati suggeriscono che i glicani di tipo complesso sono sotto gerarchie di controllo differenziale in cui alcune forme di elaborazione dei glicani potrebbero essere influenzate dalla presentazione strutturale dei siti glicani. L’attaccamento di fucosio, galattosio e acido sialico per questi siti RBD sembra essere controllato da fenomeni più globali, come la cellula produttrice, poiché l’attaccamento di questi monosaccaridi è simile quando si confrontano l’RBD monomerico e la proteina S ricombinante trimerica che sono state prodotte in linee cellulari identiche. La ramificazione dei siti RBD su RBD monomerico è maggiore di quella della proteina S trimerica,sia virale che ricombinante, e suggerisce che la struttura quaternaria della glicoproteina possa svolgere un piccolo ruolo nell’elaborazione dei glicani di tipo complesso del RBD. Questi risultati sono simili a quelli delle analisi precedenti che hanno confrontato la proteina S trimerica con la S1 monomerica. (38)
Per esplorare ulteriormente le differenze nella glicosilazione di RBD PNGS, abbiamo eseguito un’analisi comparativa simile su MERS-CoV. L’analisi sito-specifica del glicano della proteina MERS-CoV S ricombinante è stata riportata in precedenza. (51)Per l’analisi comparativa della proteina ricombinante trimerica e dell’RBD, i file MS ottenuti nello studio precedente sono stati ricercati utilizzando una versione identica del software di analisi utilizzando le stesse librerie di glicani dell’analisi mostrata nelle Figure 1 e 2 . Questa analisi ha rivelato differenze nei siti glicani presenti sul MERS RBD quando espressi monomericamente rispetto alla proteina S solubile trimerica ricombinante. Uno dei siti, N487, è simile tra le due piattaforme, presentando glicoforme tipiche di siti popolati da glicani di tipo complesso ( Figura 3B). Al contrario, N410 è occupato esclusivamente da glicani di tipo oligomannosio quando presente sulla proteina S trimerica ricombinante. Quando viene espresso il MERS RBD monomerico, il livello di elaborazione del sito N410 è notevolmente aumentato, con i glicani di tipo complesso che dominano il profilo dei glicani, sebbene rimanga una sottopopolazione di glicani di tipo oligomannosio ( Figura 3 B). La modellazione del glicano N410 su strutture pubblicate della proteina MERS S rivela che il glicano N410 è protetto dall’elaborazione da parte di enzimi mannosidasi come ER α-mannosidasi I quando sepolto all’interno del trimero, ma quando presentato sul RBD monomerico, è facilmente accessibile al glicano enzimi di elaborazione ( Figura S2). Queste osservazioni evidenziano ulteriormente come la struttura quaternaria di una glicoproteina sia un determinante chiave dello stato di elaborazione del glicano e dimostrano come i siti dei glicani possano fornire informazioni sul ripiegamento delle proteine e sulla struttura quaternaria.
Le simulazioni di dinamica molecolare rivelano la relazione tra l’accessibilità e l’elaborazione dei glicani
Utilizzando l’analisi del glicano sito-specifica, è possibile dedurre i vincoli strutturali posti sull’elaborazione del glicano di particolari PNGS dalla struttura quaternaria della proteina. Le simulazioni MD possono aiutare a capire come la flessibilità delle proteine può influenzare l’elaborazione del glicano. Le proteine ricombinanti analizzate in questo studio utilizzano tutte sostituzioni di prolina per stabilizzare la conformazione della proteina S di prefusione. Le analisi strutturali della proteina S contenente queste sostituzioni di prolina hanno mostrato che una delle tre subunità mostra frequentemente il suo RBD in una conformazione verso l’alto. (28) Eseguendo simulazioni utilizzando modelli contenenti un RBD up, è possibile studiare come una presentazione differenziale di RBD può influire sulla glicosilazione sito-specifica.
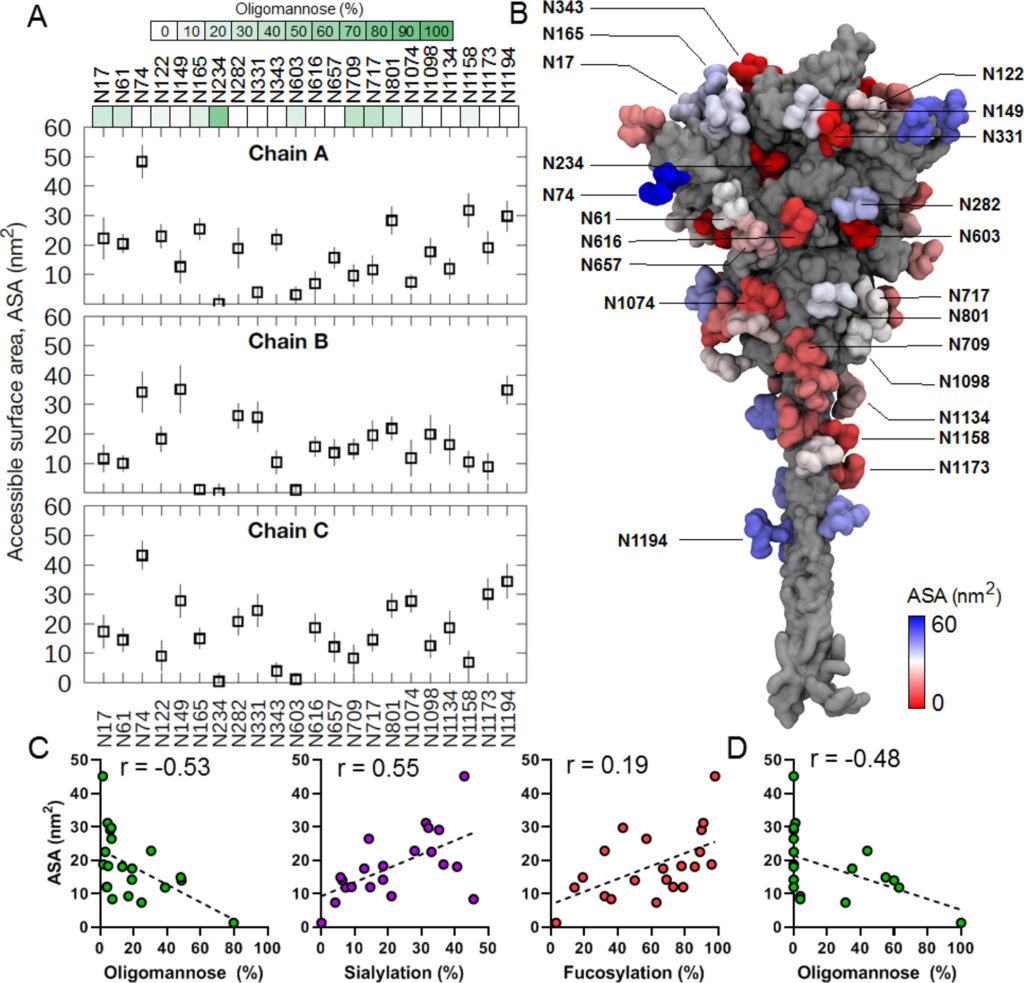
A tal fine, abbiamo eseguito simulazioni MD di 200 ns di proteina S trimerica completamente glicosilata incorporata all’interno di un modello di membrana ERGIC (dettagli in Materiali e Metodi ). Il glicano Man 9 GlcNAc 2 (Man-9), che rappresenta il substrato principale per gli enzimi di elaborazione del glicano, è stato aggiunto a ciascun PNGS per comprendere l’effetto della dinamica di proteine e glicani sull’elaborazione del glicano. L’accessibilità dei glicani agli enzimi è stata quindi chiarita calcolando l’area superficiale accessibile (ASA). Per garantire la corretta dimensione della sonda utilizzata per il calcolo dell’ASA, abbiamo prima esaminato la struttura delle mannosidasi e delle glicosiltransferasi legate al suo substrato o analogo del substrato. (91,92)Misurando la distanza tra il sito di legame del substrato e la superficie esterna degli enzimi, abbiamo scoperto che una sonda con un raggio di 1,25-1,5 nm si avvicinava al meglio alla dimensione degli enzimi ( Figura S3 ). Abbiamo misurato i valori di ASA per la catena A della proteina S utilizzando sonde con raggi di 1,25 e 1,5 nm e abbiamo scoperto che entrambi davano risultati molto simili. Tracciare i punti accessibili di una sonda da 1,5 nm attorno ai singoli glicani indicava anche che sarebbe stato necessario un raggio di 1,5 nm perché mannosidasi e glicosiltransferasi potessero accedere al glicano Man-9. Abbiamo quindi utilizzato una sonda da 1,5 nm per misurare l’ASA di ciascun glicano Man-9 sul modello della proteina S durante la simulazione MD.
Il confronto dell’ASA di ciascun glicano su ciascun protomero rivela una vasta gamma di accessibilità ( Figura 4 A,B). Alcuni siti come N234 sono stati determinati a possedere bassi valori di ASA ( Figura S4A ), indicando che questo sito è estremamente sepolto, in correlazione con le osservazioni che N234 è il meno elaborato su tutta la proteina S ricombinante e derivata da virus. Al contrario, il sito più esposto in tutte e tre le catene è N74 ( Figura S4B ). Questo glicano è altamente elaborato quando analizzato da LC-MS. È interessante notare che analisi precedenti hanno dimostrato che N74 possiede glicani solfatati. (15)Abbiamo incluso composizioni solfatate nella nostra libreria di glicani per cercare i dati MS e osservare che N74 contiene più composizioni di glicani solfatati su tutti i campioni prodotti nelle cellule HEK293 ( Tabella S1 ). La maggiore accessibilità di questo sito può spiegare perché i glicani solfatati si osservano a maggiori abbondanze in questo sito, ma non in altri. Alcuni glicani mostrano un modello di accessibilità distintamente bimodale, in cui sono stati osservati valori di ASA alti e bassi lungo la traiettoria ( Figura S5). Ad esempio, due dei glicani N122 sono stati sepolti all’interno di una fessura tra il dominio N-terminale di una catena e l’RBD di una catena adiacente durante una parte della simulazione. Allo stesso modo, uno dei glicani N603 è stato inserito in un ampio solco interprotomerico vicino al sito di scissione S1/S2. Questa osservazione è correlata con i dati LC-MS della proteina S ricombinante e derivata dal virus, mostrando che questi due siti sono popolati da circa metà oligomannosio/ibrido e metà glicani complessi, potenzialmente a causa della capacità di questi glicani di essere sepolti o esposti su la superficie proteica. Anche il glicano su N165 ha mostrato valori di ASA bimodali. Tuttavia, ciò è dovuto alla configurazione RBD up, che consente al glicano N165 di inserirsi nello spazio tra l’RBD e il dominio N-terminale, che non era accessibile nella configurazione RBD down. N165,insieme ai glicani N234, ha dimostrato di modulare la dinamica conformazionale del RBD mantenendo la configurazione up necessaria per il riconoscimento ACE2. (31)
Biochemistry 2021
Figura 4. Area superficiale accessibile (ASA) di glicani di tipo oligomannosio da simulazioni MD. (A) I valori di ASA sono stati calcolati per ciascun glicano di tipo oligomannosio (M9) per tutte e tre le catene proteiche S. In questo modello, la catena A è modellata nella conformazione RBD “up”. Gli ultimi 50 ns della simulazione sono stati utilizzati per il calcolo e le barre di errore indicano la deviazione standard lungo la traiettoria. La dimensione della sonda utilizzata era di 1,5 nm. Le barre di colore verde rappresentano il contenuto di oligomannosio dei rispettivi siti, calcolato come contenuto medio di glicani di tipo oligomannosio delle proteine S ricombinanti analizzate in questo lavoro (proteine S di Amsterdam, Harvard, Swiss e Oxford). (B) Struttura della proteina S (grigia) con glicani colorati in base ai loro valori ASA mostrati nella rappresentazione superficiale. (C) Grafici che confrontano l’elaborazione del glicano con i valori ASA calcolati,media per le catene A–C, per la proteina ricombinante media per il contenuto di glicani di tipo oligomannosio, la proporzione di glicani contenenti almeno un acido sialico e la proporzione di glicani contenenti almeno un fucosio. Il segnalatoI valori r rappresentano il coefficiente di correlazione del rango di Spearman. (D) Contenuto di glicani di tipo oligomannosio vs ASA per la proteina SARS-CoV-2 S di origine virale presentata come nel pannello C.
Per comprendere la relazione tra la presentazione di glicani di tipo oligomannoso e l’area superficiale accessibile di ciascun sito di glicano, abbiamo utilizzato il contenuto medio di glicani di tipo oligomannosio di ciascun sito come determinato nella Figura 1 , sia per il materiale ricombinante che derivato da virus, e lo abbiamo confrontato ai valori medi di ASA per le catene A–C, poiché LC-MS non è in grado di distinguere quale glicano provenga da quale catena ( Figura 4CD). Sia per la proteina S ricombinante che per quella di derivazione virale, c’era una correlazione negativa tra ASA e contenuto di oligomannosio (coefficienti di correlazione di Spearman di -0,53 per ricombinante e -0,48 per derivata virale). Ciò dimostra il legame tra l’elaborazione del glicano e l’architettura quaternaria, poiché i siti meno accessibili agli enzimi di elaborazione del glicano presentano popolazioni più grandi di glicani di tipo oligomannosio processati in modo immaturo. Al contrario, il trattamento di glicani di tipo complesso come l’aggiunta di acido sialico correlato con la superficie accessibile a livello sito-specifico ( r = 0,55) e anche fucosilazione, anche se in misura minore ( r= 0,19). Sebbene vi sia una correlazione osservata tra glicani di tipo oligomannosio e ASA, la scala temporale dei nanosecondi acquisita durante le simulazioni MD è più breve del tempo complessivo impiegato per l’elaborazione dei glicani nell’ER e nell’apparato di Golgi, e quindi la proteina S sarà per campionare più conformazioni non risolte durante la simulazione.
Infine, per determinare il potenziale effetto del raggruppamento di glicani sull’accessibilità degli enzimi e sulla successiva elaborazione dei glicani, abbiamo calcolato il numero di contatti effettuati da ciascun glicano in un dato sito con i suoi glicani circostanti durante la simulazione ( Figura S6). È interessante notare che in tutte e tre le catene i siti sulla subunità S2 mostrano un numero maggiore di contatti glicano-glicano, suggerendo che è più probabile che formino cluster locali. Una sovrapposizione di istantanee di simulazione consecutive di glicani mostra anche chiaramente che la regione S2 è più densamente ricca di glicani rispetto ai domini S1 e HR2. Ciò concorda con i dati LC-MS che mostrano che i siti sulla subunità S2, in particolare N709, N717 e N801, hanno un alto grado di glicani di tipo oligomannosio non processati. L’elevata densità di contatti glicano-glicano all’interno del cerotto di mannosio glicano è stata precedentemente implicata nella ridotta processività dagli enzimi di elaborazione del glicano nell’HIV-1 gp120, (93)e un meccanismo simile potrebbe spiegare l’aumento del contenuto di oligomannosio nei siti S2 di SARS-CoV-2. Uno studio recente ha mostrato che gli anticorpi Fab-dimerizzati che legano i glicani riconoscono i glicani di tipo oligomannosio sottotrattati su SARS-CoV-2 tramite un epitopo comune intorno a N709 sulla subunità S2, (94)suggerendo ulteriormente la formazione di un cluster glicano locale. Nel complesso, il nostro campionamento di simulazione MD mostra che l’accessibilità del glicano agli enzimi ospiti, che è influenzata dalla struttura quaternaria della proteina e dalla densità locale del glicano, è un importante determinante dell’elaborazione del glicano.
L’impatto globale di COVID-19 ha portato i laboratori di tutto il mondo a produrre proteine spike ricombinanti per la progettazione di vaccini, test antigenici e caratterizzazione strutturale. Mentre il sito di attacco dei glicani è codificato dal genoma virale, l’elaborazione dei glicani attaccati può anche essere influenzata da un’ampia gamma di fenomeni esogeni, inclusi sistemi ospiti ricombinanti e processi per la produzione in coltura cellulare. Utilizzando materiali immediatamente disponibili, abbiamo confrontato la glicosilazione delle preparazioni virali prodotte da Vero per le proteine spike con quelle delle proteine spike ricombinanti HEK-293 e derivate da CHO. Siamo consapevoli che queste preparazioni possono essere diverse dalle proteine spike prodotte in una varietà di cellule in pazienti infetti. Tuttavia,qui mostriamo che quando un sito di glicano si trova in regioni della proteina SARS-CoV-2 S che non sono facilmente accessibili, il sito di glicano possiederà alti livelli di glicani di tipo oligomannosio sottoelaborati, un fenomeno che sarà probabilmente di natura generale . Questa conclusione deriva dall’osservazione che la sottoelaborazione dei glicani si trova nei preparati proteici di una serie di istituzioni in tutto il mondo e sono presenti anche sulla proteina S derivata dalle cellule Vero derivata dalla SARS-CoV-2 infettiva. Mentre la presenza di glicani di tipo oligomannosio è rara sulla maggior parte delle glicoproteine dell’ospite, la loro abbondanza su SARS-CoV-2 è inferiore a quella di altre glicoproteine virali e significa che la densità dello scudo glicano di questo virus è bassa rispetto a quella dell’HIV -1 Env e Lassa GPC.È probabile che ciò significhi che gli epitopi della proteina immunodominante rimangono esposti. L’equivalenza di elaborazione tra proteina S ricombinante e derivata viralmente indica che la glicosilazione della proteina S ricombinante per le vaccinazioni probabilmente imiterà quelle che si verificano nelle infezioni umane e rimarrà antigenicamente comparabile. Analizzando l’elaborazione di glicani di tipo complesso su più campioni, dimostriamo che questa glicosilazione è guidata più da altri parametri, tra cui la cellula produttrice e le condizioni di coltura. Un’eccezione osservata a questo è stata che l’espressione di RBD monomerico rispetto alla proteina S trimerica aumenta il livello di ramificazione del glicano sui due siti di glicani situati sul RBD SARS-CoV-2. Ciò suggerisce che il trattamento dei glicani di glicani di tipo complesso, oltre ai glicani di tipo oligomannosio,possono anche essere soggetti a vincoli strutturali, anche se in misura molto minore. Nel complesso, questi risultati dimostrano che mentre la glicosilazione legata all’N è molto diversificata in alcune regioni della proteina S, esiste un ampio consenso sull’elaborazione dei glicani per quanto riguarda i glicani di tipo oligomannosio tra virus e proteina S dell’immunogeno. Questo è qualcosa che non può essere dato per scontato, poiché al confronto di HIV-1 Env ricombinante e di derivazione virale, la ridotta occupazione di glicani degli immunogeni può indurre una risposta immunitaria incapace di neutralizzare il virus. La riproducibilità della glicosilazione della proteina S da molte fonti diverse è di notevole beneficio per la progettazione dell’immunogeno, i test sierologici e la scoperta di farmaci e significherà che è improbabile che i glicani di SARS-CoV-2 forniscano una barriera alla lotta contro la pandemia di COVID-19.anche se in misura molto minore. Nel complesso, questi risultati dimostrano che mentre la glicosilazione legata all’N è molto diversificata in alcune regioni della proteina S, esiste un ampio consenso sull’elaborazione dei glicani per quanto riguarda i glicani di tipo oligomannosio tra virus e proteina S dell’immunogeno. Questo è qualcosa che non può essere dato per scontato, poiché al confronto di HIV-1 Env ricombinante e di derivazione virale, la ridotta occupazione di glicani degli immunogeni può indurre una risposta immunitaria incapace di neutralizzare il virus. La riproducibilità della glicosilazione della proteina S da molte fonti diverse è di notevole beneficio per la progettazione dell’immunogeno, i test sierologici e la scoperta di farmaci e significherà che è improbabile che i glicani di SARS-CoV-2 forniscano una barriera alla lotta contro la pandemia di COVID-19.anche se in misura molto minore. Nel complesso, questi risultati dimostrano che mentre la glicosilazione legata all’N è molto diversificata in alcune regioni della proteina S, esiste un ampio consenso sull’elaborazione dei glicani per quanto riguarda i glicani di tipo oligomannosio tra virus e proteina S dell’immunogeno. Questo è qualcosa che non può essere dato per scontato, poiché al confronto di HIV-1 Env ricombinante e di derivazione virale, la ridotta occupazione di glicani degli immunogeni può indurre una risposta immunitaria incapace di neutralizzare il virus. La riproducibilità della glicosilazione della proteina S da molte fonti diverse è di notevole beneficio per la progettazione dell’immunogeno, i test sierologici e la scoperta di farmaci e significherà che è improbabile che i glicani di SARS-CoV-2 forniscano una barriera alla lotta contro la pandemia di COVID-19.questi risultati dimostrano che mentre la glicosilazione legata all’N è molto diversificata in alcune regioni della proteina S, esiste un ampio consenso sull’elaborazione dei glicani per quanto riguarda i glicani di tipo oligomannosio tra virus e proteina S dell’immunogeno. Questo è qualcosa che non può essere dato per scontato, poiché al confronto di HIV-1 Env ricombinante e di derivazione virale, la ridotta occupazione di glicani degli immunogeni può indurre una risposta immunitaria incapace di neutralizzare il virus. La riproducibilità della glicosilazione della proteina S da molte fonti diverse è di notevole beneficio per la progettazione dell’immunogeno, i test sierologici e la scoperta di farmaci e significherà che è improbabile che i glicani di SARS-CoV-2 forniscano una barriera alla lotta contro la pandemia di COVID-19.questi risultati dimostrano che mentre la glicosilazione legata all’N è molto diversificata in alcune regioni della proteina S, esiste un ampio consenso sull’elaborazione dei glicani per quanto riguarda i glicani di tipo oligomannosio tra virus e proteina S dell’immunogeno. Questo è qualcosa che non può essere dato per scontato, poiché al confronto di HIV-1 Env ricombinante e di derivazione virale, la ridotta occupazione di glicani degli immunogeni può indurre una risposta immunitaria incapace di neutralizzare il virus. La riproducibilità della glicosilazione della proteina S da molte fonti diverse è di notevole beneficio per la progettazione dell’immunogeno, i test sierologici e la scoperta di farmaci e significherà che è improbabile che i glicani di SARS-CoV-2 forniscano una barriera alla lotta contro la pandemia di COVID-19.vi è un ampio consenso sull’elaborazione dei glicani per quanto riguarda i glicani di tipo oligomannosio tra virus e proteina S dell’immunogeno. Questo è qualcosa che non può essere dato per scontato, poiché al confronto di HIV-1 Env ricombinante e di derivazione virale, la ridotta occupazione di glicani degli immunogeni può indurre una risposta immunitaria incapace di neutralizzare il virus. La riproducibilità della glicosilazione della proteina S da molte fonti diverse è di notevole beneficio per la progettazione dell’immunogeno, i test sierologici e la scoperta di farmaci e significherà che è improbabile che i glicani di SARS-CoV-2 forniscano una barriera alla lotta contro la pandemia di COVID-19.esiste un ampio consenso sull’elaborazione dei glicani per quanto riguarda i glicani di tipo oligomannosio tra virus e proteina S dell’immunogeno. Questo è qualcosa che non può essere dato per scontato, poiché al confronto di HIV-1 Env ricombinante e di derivazione virale, la ridotta occupazione di glicani degli immunogeni può indurre una risposta immunitaria incapace di neutralizzare il virus. La riproducibilità della glicosilazione della proteina S da molte fonti diverse è di notevole beneficio per la progettazione dell’immunogeno, i test sierologici e la scoperta di farmaci e significherà che è improbabile che i glicani di SARS-CoV-2 forniscano una barriera alla lotta contro la pandemia di COVID-19.come nel confronto tra HIV-1 Env ricombinante e di derivazione virale, la ridotta occupazione di glicani degli immunogeni può indurre una risposta immunitaria incapace di neutralizzare il virus. La riproducibilità della glicosilazione della proteina S da molte fonti diverse è di notevole beneficio per la progettazione dell’immunogeno, i test sierologici e la scoperta di farmaci e significherà che è improbabile che i glicani di SARS-CoV-2 forniscano una barriera alla lotta contro la pandemia di COVID-19.come nel confronto tra HIV-1 Env ricombinante e di derivazione virale, la ridotta occupazione di glicani degli immunogeni può indurre una risposta immunitaria incapace di neutralizzare il virus. La riproducibilità della glicosilazione della proteina S da molte fonti diverse è di notevole beneficio per la progettazione dell’immunogeno, i test sierologici e la scoperta di farmaci e significherà che è improbabile che i glicani di SARS-CoV-2 forniscano una barriera alla lotta contro la pandemia di COVID-19.e scoperta di farmaci e significherà che è improbabile che i glicani di SARS-CoV-2 forniscano una barriera alla lotta contro la pandemia di COVID-19.e scoperta di farmaci e significherà che è improbabile che i glicani di SARS-CoV-2 forniscano una barriera alla lotta contro la pandemia di COVID-19.
Average recombinant and virally derived S protein site-specific glycan compositions averaged (Table S1), percentage point change in glycosylation on virally derived S protein compared to recombinant S protein (Table S2), site-specific glycan compositions of the two RBD sites, N331 and N343 (Table S3), BLAST alignment of recombinant S proteins (Figure S1), model demonstrating the differential accessibility of the N410 glycan for glycan processing enzymes between trimeric MERS S protein and the monomeric MERS RBD (Figure S2), validation of probe size for ASA measurement (Figure S3), examples of glycans either buried or exposed in all three chains (Figure S4), examples of glycans with bimodal accessibility properties (Figure S5), and glycan–glycan contacts from MD simulations (Figure S6) (PDF)
Accession Codes
Spike glycoprotein, UniProtKB P0DTC2 (SPIKE_SARS2); MERS CoV Spike glycoprotein, UniProtKB R9UQ53 (R9UQ53_MERS); raw file accession, MassIVE MSV000087308.
Corresponding Authors
Peter J. Bond – Bioinformatics Institute, Agency for Science, Technology and Research (A*STAR), Singapore 138671; Department of Biological Sciences, National University of Singapore, Singapore 117543; https://orcid.org/0000-0003-2900-098X; Email: peterjb@bii.a-star.edu.sg
Firdaus Samsudin – Bioinformatics Institute, Agency for Science, Technology and Research (A*STAR), Singapore 138671
Lorena Zuzic – Bioinformatics Institute, Agency for Science, Technology and Research (A*STAR), Singapore 138671; Department of Chemistry, Faculty of Science and Engineering, Manchester Institute of Biotechnology, The University of Manchester, Manchester M1 7DN, U.K.; https://orcid.org/0000-0002-7834-612X
Aishwary Tukaram Shivgan – Bioinformatics Institute, Agency for Science, Technology and Research (A*STAR), Singapore 138671; Department of Biological Sciences, National University of Singapore, Singapore 117543; https://orcid.org/0000-0002-2032-8738
Wan-ting He – Department of Immunology and Microbiology, The Scripps Research Institute, La Jolla, California 92037, United States; IAVI Neutralizing Antibody Center, The Scripps Research Institute, La Jolla, California 92037, United States; Consortium for HIV/AIDS Vaccine Development (CHAVD), The Scripps Research Institute, La Jolla, California 92037, United States
Sean Callaghan – Department of Immunology and Microbiology, The Scripps Research Institute, La Jolla, California 92037, United States; IAVI Neutralizing Antibody Center, The Scripps Research Institute, La Jolla, California 92037, United States; Consortium for HIV/AIDS Vaccine Development (CHAVD), The Scripps Research Institute, La Jolla, California 92037, United States
Ge Song – Department of Immunology and Microbiology, The Scripps Research Institute, La Jolla, California 92037, United States; IAVI Neutralizing Antibody Center, The Scripps Research Institute, La Jolla, California 92037, United States; Consortium for HIV/AIDS Vaccine Development (CHAVD), The Scripps Research Institute, La Jolla, California 92037, United States
Peter Yong – Department of Immunology and Microbiology, The Scripps Research Institute, La Jolla, California 92037, United States; IAVI Neutralizing Antibody Center, The Scripps Research Institute, La Jolla, California 92037, United States; Consortium for HIV/AIDS Vaccine Development (CHAVD), The Scripps Research Institute, La Jolla, California 92037, United States; https://orcid.org/0000-0002-4899-3270
Philip J. M. Brouwer – Department of Medical Microbiology, Amsterdam UMC, University of Amsterdam, Amsterdam Infection & Immunity Institute, 1007 MB Amsterdam, The Netherlands
Yutong Song – Tsinghua-Peking Center for Life Sciences, School of Life Sciences, Tsinghua University, Beijing 100084, China; Beijing Advanced Innovation Center for Structural Biology and Frontier Research Center for Biological Structure, Beijing 100084, China; https://orcid.org/0000-0002-2206-9549
Yongfei Cai – Division of Molecular Medicine, Boston Children’s Hospital, 3 Blackfan Street, Boston, Massachusetts 02115, United States
Helen M. E. Duyvesteyn – Division of Structural Biology, University of Oxford, The Wellcome Centre for Human Genetics, Headington, Oxford OX3 7BN, U.K.
Tomas Malinauskas – Division of Structural Biology, University of Oxford, The Wellcome Centre for Human Genetics, Headington, Oxford OX3 7BN, U.K.; https://orcid.org/0000-0002-4847-5529
Joeri Kint – ExcellGene SA, CH1870 Monthey, Switzerland
Paco Pino – ExcellGene SA, CH1870 Monthey, Switzerland
Maria J. Wurm – ExcellGene SA, CH1870 Monthey, Switzerland
Bing Chen – Division of Molecular Medicine, Boston Children’s Hospital, 3 Blackfan Street, Boston, Massachusetts 02115, United States; Department of Pediatrics, Harvard Medical School, 3 Blackfan Street, Boston, Massachusetts 02115, United States
David I. Stuart – Division of Structural Biology, University of Oxford, The Wellcome Centre for Human Genetics, Headington, Oxford OX3 7BN, U.K.; Diamond Light Source Ltd., Harwell Science & Innovation Campus, Didcot OX11 0DE, U.K.; https://orcid.org/0000-0002-3426-4210
Rogier W. Sanders – Department of Medical Microbiology, Amsterdam UMC, University of Amsterdam, Amsterdam Infection & Immunity Institute, 1007 MB Amsterdam, The Netherlands; Department of Microbiology and Immunology, Weill Medical College of Cornell University, New York, New York 10065, United States
Raiees Andrabi – Department of Immunology and Microbiology, The Scripps Research Institute, La Jolla, California 92037, United States; IAVI Neutralizing Antibody Center, The Scripps Research Institute, La Jolla, California 92037, United States; Consortium for HIV/AIDS Vaccine Development (CHAVD), The Scripps Research Institute, La Jolla, California 92037, United States
Dennis R. Burton – Department of Immunology and Microbiology, The Scripps Research Institute, La Jolla, California 92037, United States; IAVI Neutralizing Antibody Center, The Scripps Research Institute, La Jolla, California 92037, United States; Consortium for HIV/AIDS Vaccine Development (CHAVD), The Scripps Research Institute, La Jolla, California 92037, United States; Ragon Institute of Massachusetts General Hospital, Massachusetts Institute of Technology, and Harvard University, Cambridge, Massachusetts 02139, United States
Sai Li – Tsinghua-Peking Center for Life Sciences, School of Life Sciences, Tsinghua University, Beijing 100084, China; Beijing Advanced Innovation Center for Structural Biology and Frontier Research Center for Biological Structure, Beijing 100084, China; https://orcid.org/0000-0002-9353-0355
Author ContributionsJ.D.A., H.C., and F.S. contributed equally to this work. J.D.A., Y.W., and H.C. performed site-specific analysis. J.D.A., F.S., H.C., P.J.B., and M.C. wrote and edited the manuscript. F.S., L.Z., A.T.S., M.F., and P.J.B. performed MD simulations. W.H., S.C., G.S., P.Y., R.A., and D.R.B. provided monomeric RBD proteins. P.J.M.B., Y.C., H.M.E.D., T.M., J.K., P.P., M.J.W., B.C., D.I.S., and R.W.S. provided recombinant S protein. Y.S. and S.L. produced and collected LC-MS data for viral derived S protein. All authors edited and reviewed the manuscript prior to submission.
NotesThe authors declare the following competing financial interest(s): ExcellGene sells purified trimeric spike protein preparations from CHO cells to commercial companies for internal research and for use in diagnostic applications.